

















 89 / 103
89 / 103

Page 128
Notes:
conferenceseries
.com
Volume 10, Issue 8 (Suppl)
J Proteomics Bioinform, an open access journal
ISSN: 0974-276X
Structural Biology 2017
September 18-20, 2017
9
th
International Conference on
Structural Biology
September 18-20, 2017 Zurich, Switzerland
Paola Picotti, J Proteomics Bioinform 2017, 10:8(Suppl)
DOI: 10.4172/0974-276X-C1-0100
Monitoring protein structural changes on a proteome-wide scale
Paola Picotti
ETH Zurich, Switzerland
P
rotein structural changes induced by external perturbations or internal cues can profoundly influence protein activity and
thus modulate cellular physiology. Mass spectrometry (MS)-based proteomic techniques are routinely used to measure
changes in protein abundance, post-translational modification and protein interactors, but much less is known about protein
structural changes, owing to the lack of suitable approaches to study global changes in protein folds in cells. In my talk, I
will present a novel structural proteomics technology developed by our group that enables the analysis of protein structural
changes on a proteome-wide scale and directly in complex biological extracts. The approach relies on the coupling of limited
proteolysis (LiP) tools and an advanced MS workflow. LiP-MS can detect subtle alterations in secondary structure content,
larger scale movements such as domain motions, and more pronounced transitions such as the switch between folded and
unfolded states or multimerization events. The method can also be used to pinpoint protein regions undergoing a structural
transition with peptide-level resolution. I will describe selected applications of the approach, including 1. The identification of
proteins that undergo structural rearrangements in cells due to a nutrient shift; 2. The analysis of
in vivo
protein aggregation;
3. The cell-wide analysis of protein thermal unfolding; and 4. The identification of protein-small molecule interactions (e.g.
drug-target deconvolution). I will discuss the power and limitations of the method and possible new directions in structural
biology enabled by this emerging approach to protein structure analysis.
Biography
Paola Picotti completed her PhD at the University of Padua (Italy) and then joined the group of Ruedi Aebersold at ETH Zurich (Switzerland), where she developed
novel targeted proteomic techniques. In 2011, she was appointed Assistant Professor at ETH Zurich. Her group develops structural and chemoproteomics methods
and uses them to study the consequences of intracellular protein aggregation. Paola Picotti’s research was awarded an ERC Starting grant, a Professorship
grant from the Swiss National Science Foundation, the Latsis Prize, the Robert J Cotter Award, the SGMS Award and the EMBO Young Investigator Award. Main
contributions of her group are the development of a structural method to analyze protein conformational changes on a system-wide level, the discovery of novel
allosteric interactions, the analysis of the determinants of proteome thermostability and the identification of a novel neuronal clearance mechanism for a protein
involved in Parkinson’s disease.
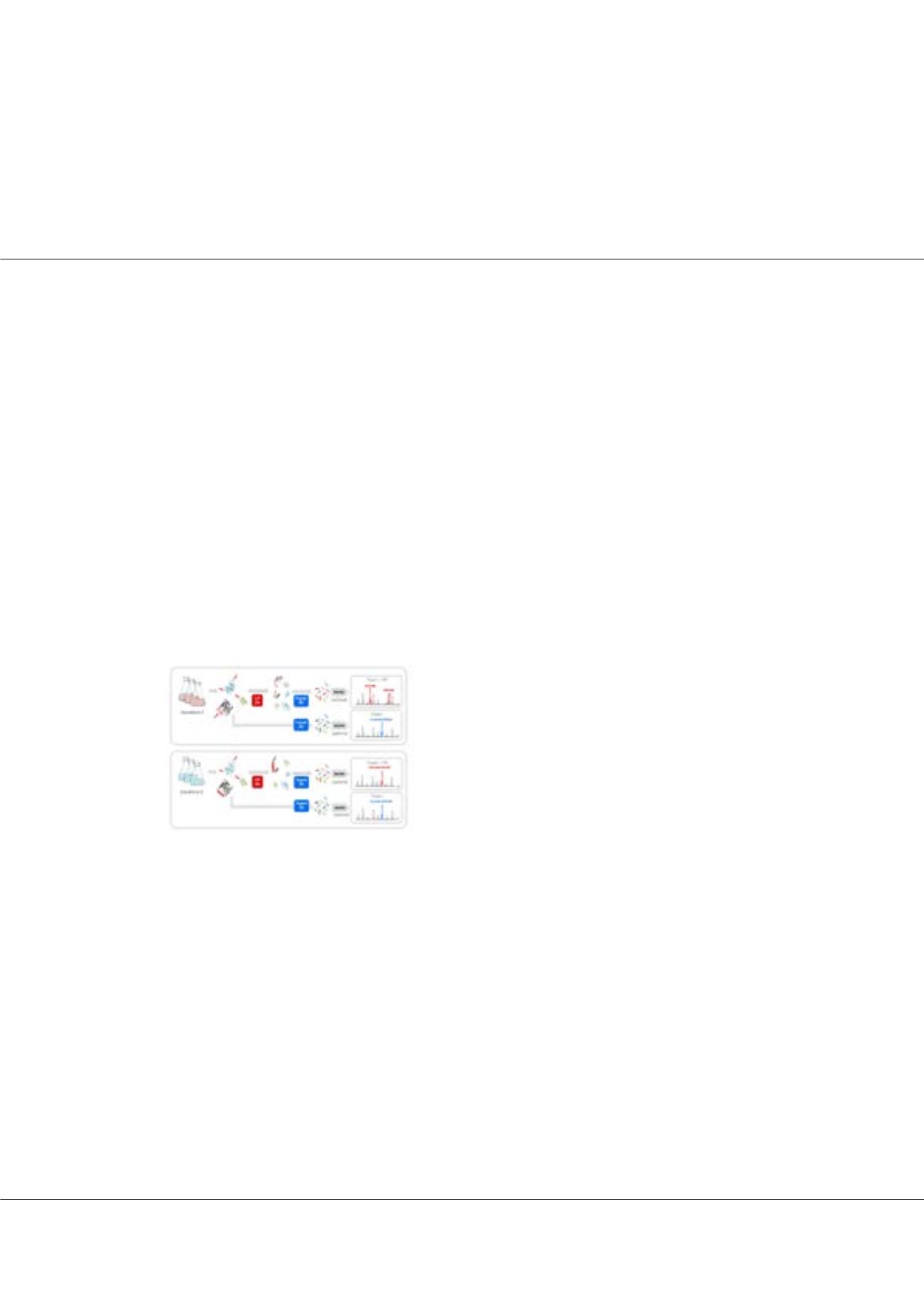
paola.picotti@bc.biol.ethz.chFigure1:
LiP-MS approach. A native proteome extract is incubated with
a broad-specificity protease for a short time. Red arrows indicate LiP
sites. Protease activity is quenched by shifting the proteome to denaturing
conditions and complete trypsin digestion is then performed. A fraction of
the same sample is only subjected to the trypsin step. The samples are
analyzed by MS and levels of the resulting fully tryptic (FT) and half-tryptic
(HT) peptides are compared. A FT peptide containing a LiP cleavage
site will be detected in the trypsin control and replaced by two HT halves
in the sample subjected to LiP. N and D indicate native and denaturing
conditions, respectively.