

















 7 / 103
7 / 103

Volume 10, Issue 8 (Suppl)
J Proteomics Bioinform, an open access journal
ISSN: 0974-276X
Structural Biology 2017
September 18- 20, 2017
Page 38
conference
series
.com
9
th
International Conference on
Structural Biology
September 18-20, 2017 Zurich, Switzerland
Dmitrii V Shalashilin, J Proteomics Bioinform 2017, 10:8(Suppl)
DOI: 10.4172/0974-276X-C1-0100
New applications of boxed molecular dynamics: Atomistic simulations of atomic force microscopy
experiments and peptide cyclization
N
ew applications of Boxed Dynamics (BXD), an efficient technique to extend the time scale of molecular dynamics and
simulate rare events, will be presented. BXD allows analysis of thermodynamics and kinetics in complicated molecular
systems. It is a fully atomistic multiscale technique, in which thermodynamics and long-time dynamics are recovered from
a set of short-time molecular dynamics simulations. BXD is many orders of magnitude faster than standard MD and can
produce well converged results. Previously BXD has been applied to peptide cyclization, solution-phase organic reaction
dynamics, and desorption of ions from self-assembled monolayers (SAMs). Here two new applications of BXDwill be reported.
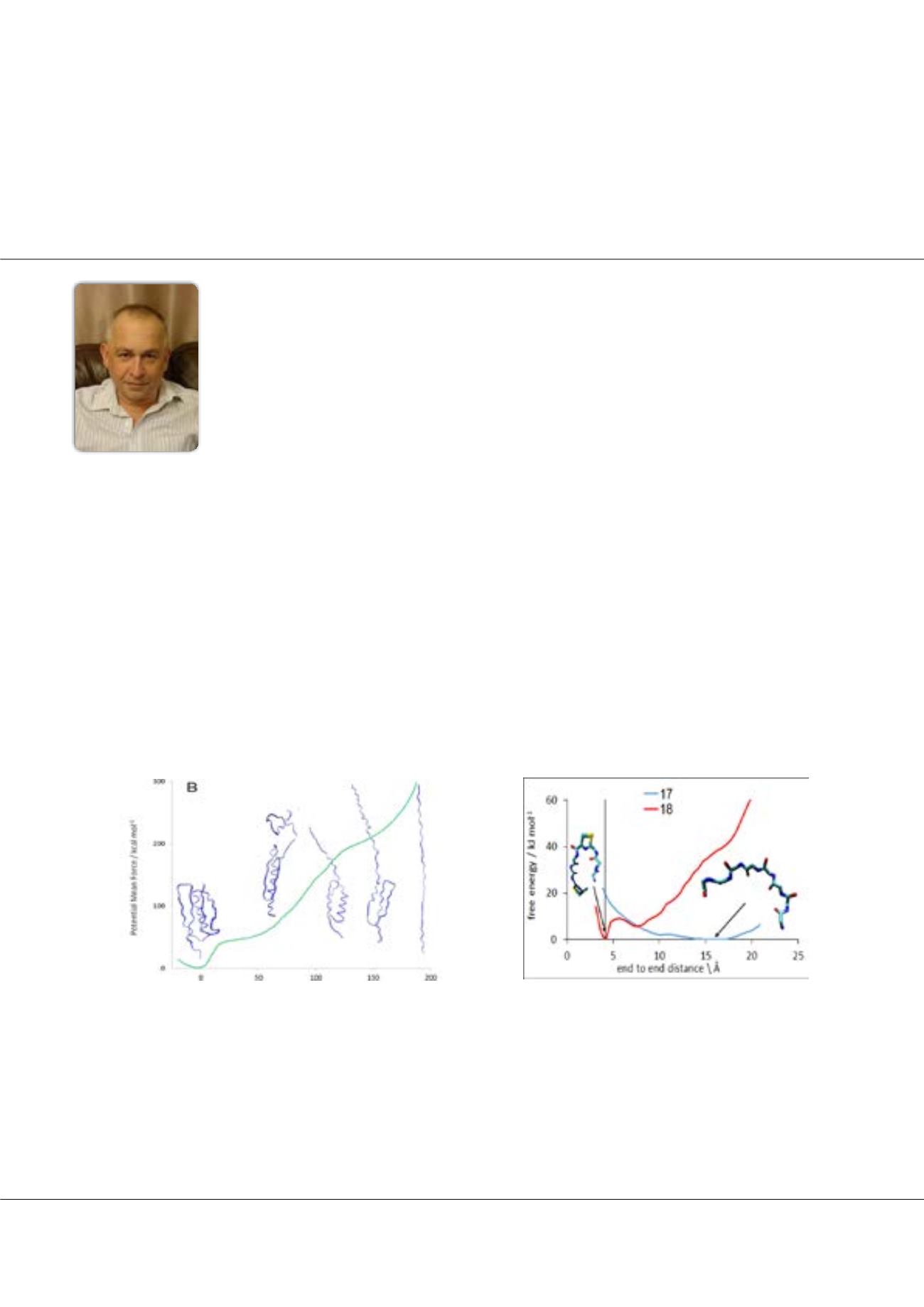
First atomistic simulations of protein pulling with Atomic Force Microscope AFM) will be presented, where BXD is able to
reproduce correctly the Potential of Mean Force (PMF) of a protein pulled in AFM experiments, the experimentally observed
force profile and its relationship with the protein structure. Second, an application of BXD to enzymatic peptide cyclization
will also be presented, where BXD predicts correctly the cyclizable peptide sequences. All such sequences have a conformation
with their C and N termini close to each other. In both applications calculations were done with standard force field without
any adjustment of the force field parameters. Thus, BXD proves to be a good predictive tool. It is implemented in CHARMM
molecular dynamics code and can be used for many other applications.
Biography
Dmitrii V Shalashilin is a Professor of Computational Chemistry at the University of Leeds. His research is focused on the development of efficient computational tech-
niques for quantum and classical simulations in chemistry and their applications.
d.shalashilin@leeds.ac.ukDmitrii V Shalashilin
University of Leeds, UK
Figure1: Potential of mean force as a function of end-to-end distance cal-
culated with BXD correlates with the structures of the unfolding protein.
Figure2: PMF as a function of end-to-end distance for two peptides
P18 and P17. Only P18, which has a stable conformation with C
and N termini close to each other, is cyclizable.