 Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi Our Group organises 3000+ Global Conferenceseries Events every year across USA, Europe & Asia with support from 1000 more scientific Societies and Publishes 700+ Open Access Journals which contains over 50000 eminent personalities, reputed scientists as editorial board members.
Open Access Journals gaining more Readers and Citations
700 Journals and 15,000,000 Readers Each Journal is getting 25,000+ Readers
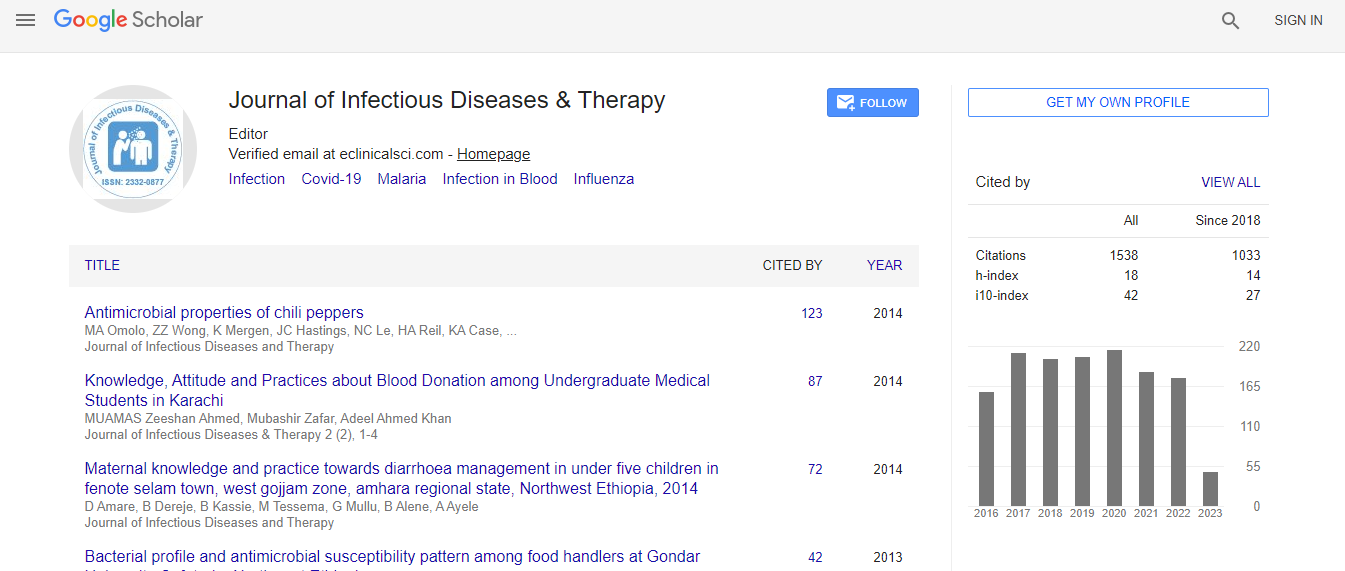
Google Scholar citation report
Citations : 1529
Journal of Infectious Diseases & Therapy received 1529 citations as per Google Scholar report
Indexed In
- Index Copernicus
- Google Scholar
- Open J Gate
- RefSeek
- Hamdard University
- EBSCO A-Z
- OCLC- WorldCat
- Publons
- Euro Pub
- ICMJE
Useful Links
Recommended Journals
Related Subjects
Share This Page
Whole genome sequencing for identification of pathogens from whole blood: Febrile neutropenia and beyond
International Conference on Infectious Diseases, Diagnostic Microbiology & Dermatologists Summit on Skin Infections
George Somerset Watts
University of Arizona Cancer Center, USA
ScientificTracks Abstracts: J Infect Dis Ther
Abstract
Febrile neutropenia (FN) is a serious complication in hematopoietic stem cell transplantation (HSCT) arising from opportunistic infections of bacterial, viral and fungal origin. FN is a major cause of morbidity, mortality and healthcare resource use in HSCT patients. Empiric antibiotic therapy is started as soon as possible once FN is recognized, accompanied by blood culture to guide rational therapy. Unfortunately, the culture-positive rate is 20% in FN patients, resulting in ineffective or sub-optimal therapy in many cases. With the high incidence of FN in HSCT patients (85-95%), HSCT patients are disproportionately affected by FN morbidity and mortality (9-14%). To improve the diagnosis of FN, we have developed an approach that utilizes whole genome sequencing to identify pathogens from whole blood samples. Here we present our initial results from HSCT patient samples taken before, during and after FN episodes. In patient samples, total reads ranged between 762465 and 18 million, with non-human reads constituting 2% of total when infection was not suspected and 14% of total when infection was suspected. A likely causative organism was identified in 82% of suspected infections. Organisms detected include Pseudomonas fluorescens, Human Parvovirus, TT virus, Escherichia coli and Enterococcus cloacae. Certain organisms were consistently found at low levels in patient and control samples, providing insight into the background contamination to be expected in this type of analysis. Our approach to identifying organisms in whole blood samples from HSCT patients with and without neutropenic fever shows promise to improve the rate of diagnosis when infection is suspected.Biography
George Somerset Watts has completed his PhD and Post-doctoral studies at the University of Arizona, USA. He has developed and Co-Directs the Genomics Core Service at the University of Arizona Cancer Center where he has performed genomics services based on microarray and sequencing technology for the past 16 years. He has published more than 25 papers in cancer research and has recently developed methods for identification of pathogens from whole blood and other biological samples which could improve management of infection in applications from diabetic ulcers to febrile neutropenia.
Email: gwatts@email.arizona.edu