 Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi Our Group organises 3000+ Global Conferenceseries Events every year across USA, Europe & Asia with support from 1000 more scientific Societies and Publishes 700+ Open Access Journals which contains over 50000 eminent personalities, reputed scientists as editorial board members.
Open Access Journals gaining more Readers and Citations
700 Journals and 15,000,000 Readers Each Journal is getting 25,000+ Readers
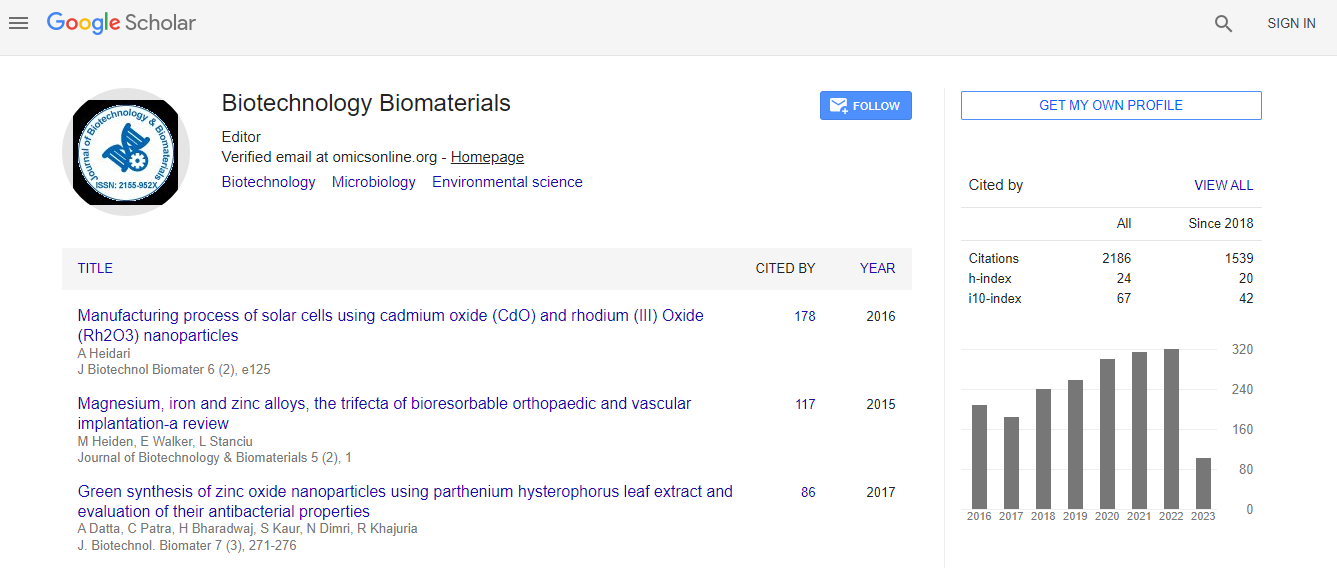
Google Scholar citation report
Citations : 3330
Journal of Biotechnology & Biomaterials received 3330 citations as per Google Scholar report
Indexed In
- Index Copernicus
- Google Scholar
- Sherpa Romeo
- Open J Gate
- Genamics JournalSeek
- Academic Keys
- ResearchBible
- China National Knowledge Infrastructure (CNKI)
- Access to Global Online Research in Agriculture (AGORA)
- Electronic Journals Library
- RefSeek
- Hamdard University
- EBSCO A-Z
- OCLC- WorldCat
- SWB online catalog
- Virtual Library of Biology (vifabio)
- Publons
- Geneva Foundation for Medical Education and Research
- Euro Pub
- ICMJE
Useful Links
Recommended Journals
Related Subjects
Share This Page
In Association with
Towards a better understanding of the biochemical response to stressors such as disease or toxins: IROA measurement system and workflow
Joint Event on 24th Biotechnology Congress: Research & Innovations & Annual Congress on CRISPR Cas9 Technology and Genetic Engineering
Felice A de Jong
IROA Technologies LLC, USA
ScientificTracks Abstracts: J Biotechnol Biomater
Abstract
Statement of the Problem: Metabolites such as glucose respond to stressors that disrupt homeostasis, such as toxins. Humans produce 1000’s of metabolites and the measurement of these molecules provides a treasure-trove of information that can be used to elucidate the health status of an organism. The measurement of metabolites (metabolomics) is widely achieved using liquid chromatography-mass spectrometry (LC-MS) whereby molecules are charged or ionized, fragmented and separated to generate peaks for 100s of metabolites from a single sample, i.e. a drop of blood. Advances in mass spectrometry, including accurate mass and high resolution, have enabled the measurement of many metabolites but still, over 60% of the data generated remains a mystery. One bottleneck is piecing together fragments with their parent molecules to achieve accurate identification of as many metabolites as possible. Confounding the issue is that most unknown peaks arise from artefacts including contaminants. The purpose of this study is to describe a QC IROA metabolomics workflow that removes artefactual peaks and increases accurate metabolite identification. Methodology & Theoretical Orientation: Two sets of 100’s of stable-labeled internal standards were generated; one enriched in one isotope and depleted in its other isotopic form, and the other its mirror image (95% 13C6 and 5% 12C6 and 5% 13C6 and 95% 12C6) so that all of the molecules in each standard exhibited unique mass spectral patterns, all mathematically calculable and easily characterized by Clusterfinder software algorithms. The paired internal standards (mixed 1:1; “Matrix”) was analyzed by LC-MS and ClusterFinder to generate an accurate library of compound and peak identifiers, including mass and retention time. The heavier isotopic standard (95% 13C6) was spiked into samples and analyzed by LC-MS. The Matrix was analyzed every ten samples to account for fluctuations in mass and retention time drift during the run. ClusterFinder located the labeled mass spectral peaks in the heavy standard and then located, identified and quantitated matching native (unlabeled) metabolite peaks in samples using the library. Peak correlation was performed to pair fragments and parent peaks. An example of this technology will be shown that illustrates the toxicological mechanism of action of flucytosine in yeast. Conclusion & Significance: Metabolite identification and quantitation are achieved using a reproducible QC workflow enabling accurate results which can be used to interrogate living systems.Biography
Felice de Jong has been an active leader and proponent in the field of metabolomics since its emergence, initially during her 6 years as Senior/Director of Business Development for Metabolon and more recently as CEO and co-founder of IROA Technologies. Here she works with collaborators to develop services and products that remove bottlenecks to streamline the measurement of biological response to stressors such as disease, adverse environmental conditions, drugs, and toxins.
E-mail: felice@iroatech.com