 Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi Our Group organises 3000+ Global Conferenceseries Events every year across USA, Europe & Asia with support from 1000 more scientific Societies and Publishes 700+ Open Access Journals which contains over 50000 eminent personalities, reputed scientists as editorial board members.
Open Access Journals gaining more Readers and Citations
700 Journals and 15,000,000 Readers Each Journal is getting 25,000+ Readers
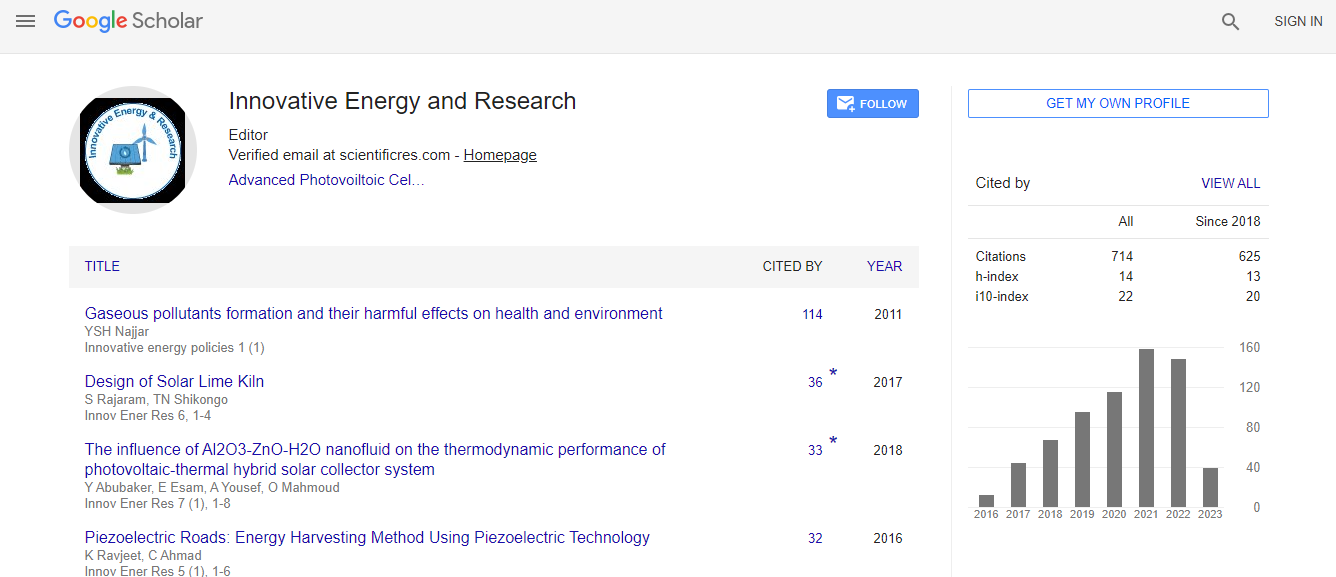
Google Scholar citation report
Citations : 712
Innovative Energy & Research received 712 citations as per Google Scholar report
Innovative Energy & Research peer review process verified at publons

Indexed In
- Google Scholar
- Open J Gate
- Genamics JournalSeek
- RefSeek
- Hamdard University
- EBSCO A-Z
- Publons
- Euro Pub
- ICMJE
Useful Links
Recommended Journals
Related Subjects
Share This Page
Elucidating the role of Sn-substitution and Pb-Vc in regulating stability and carrier concentration in CH3NH3Pb1-X-YSnXVcYI3
20th International Conference on Advanced Energy Materials and Research
Saswata Bhattacharya
Indian Institute of Technology Delhi, India
Posters & Accepted Abstracts: Innov Ener Res
Abstract
Inorganic-organic perovskites, CH3NH3PbI3 in particular, have recently drawn significant attention in improving solar cell’s performance due to their long diffusion length, high carrier mobility, suitable optical band gaps, and strong absorption of light. However, the presence of Pb has rendered its usage in developing non-toxic lead-free devices. Therefore, reducing the extent of Pb by substituting a suitable alternative metal (e.g. Sn, Ge, Sr, etc.) in the perovskite has become extremely important. Moreover, inclusion of Sn in the perovskite network (i.e. CH3NH3Pb1−xSnxI3) can reduce the optical band gap. This enables the perovskite to absorb all visible lights of the solar spectrum along with some ultraviolet to near-infrared photons (up to 1.1 eV). Therefore, digging deep into the atomistic insights and electronic structure of Sn based perovskites has become profoundly important for designing efficient energy harvesting materials. In this talk, I shall address the role of Snsubstitution and Pb-vacancy (Vc) in regulating stability and carrier concentration of CH3NH3Pb1−X−YSnXVcYI3 using state-ofthe- art density functional theory (DFT). The finite temperature effect is duly included in our theoretical calculations involving lattice thermal vibration at a given temperature. We find that at low temperature the system prefers Pb-Vc and the most stable configuration does not prefer any Pb at 50% Sn concentration. However, the Pb-Vcs become unfavourable above 250K due to the reduced linearity of I-Sn-I bonds. For n-type host the Sn substitution is more preferable than Pb-Vc formation, while for p-type host the trend is exactly opposite. The charge states of both substituted Sn and Pb-Vc are found to be dependent on the Sn concentration, which in turn alters the perovskite’s property from n-type to p-type beyond 50% Sn substitution. All our theoretical explanations, that were hitherto unknown, are perfectly in agreement with previously available experimentalobservations.Biography
E-mail: saswata@physics.iitd.ac.in