 Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi Our Group organises 3000+ Global Conferenceseries Events every year across USA, Europe & Asia with support from 1000 more scientific Societies and Publishes 700+ Open Access Journals which contains over 50000 eminent personalities, reputed scientists as editorial board members.
Open Access Journals gaining more Readers and Citations
700 Journals and 15,000,000 Readers Each Journal is getting 25,000+ Readers
Google Scholar citation report
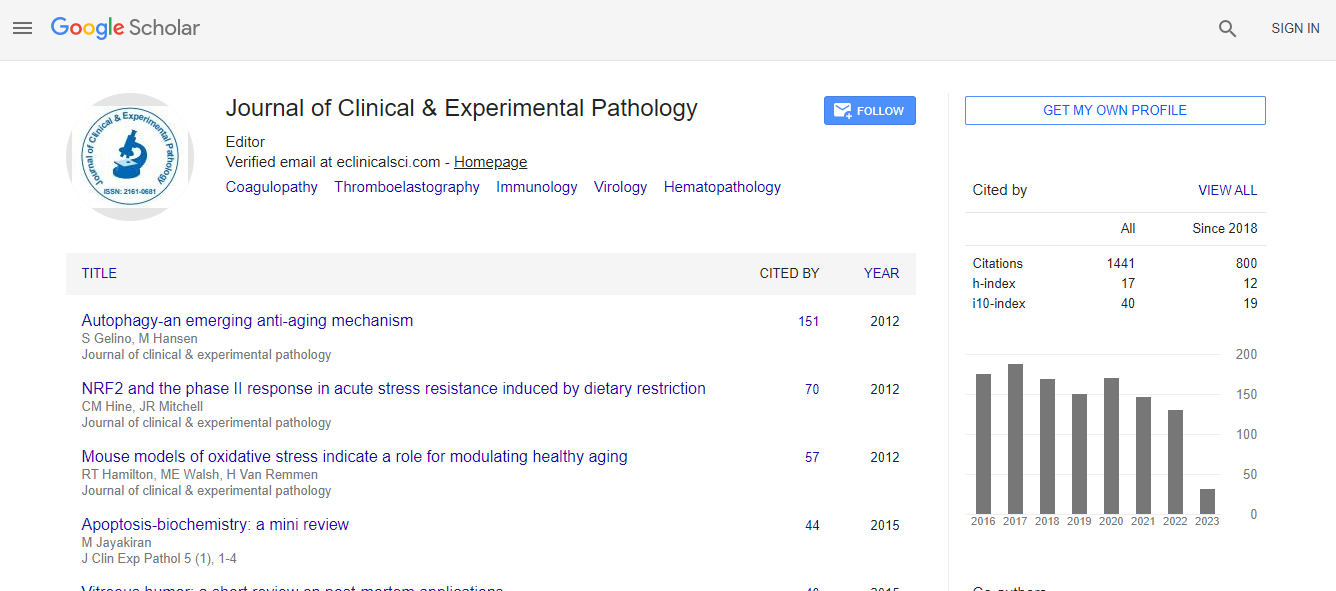
Citations : 1437
Journal of Clinical & Experimental Pathology received 1437 citations as per Google Scholar report
Journal of Clinical & Experimental Pathology peer review process verified at publons

Indexed In
- Index Copernicus
- Google Scholar
- Sherpa Romeo
- Open J Gate
- Genamics JournalSeek
- JournalTOCs
- Cosmos IF
- Ulrich's Periodicals Directory
- RefSeek
- Directory of Research Journal Indexing (DRJI)
- Hamdard University
- EBSCO A-Z
- OCLC- WorldCat
- Publons
- Geneva Foundation for Medical Education and Research
- Euro Pub
- ICMJE
- world cat
- journal seek genamics
- j-gate
- esji (eurasian scientific journal index)
Useful Links
Recommended Journals
Related Subjects
Share This Page
Autopsy findings in RMND1-related mitochondrial cytopathy
5th International Conference on Pathology
Noorah Almadani, Glenda Hendson, Hilary Valance and Jefferson Terry
University of British Columbia, Canada King Abdulaziz Medical City, KSA Children��?s and Women��?s Health Centre of British Columbia, Canada
Posters & Accepted Abstracts: J Clin Exp Pathol
Abstract
Introduction: RMND1 encodes a protein that localizes to the inner mitochondrial membrane where it plays a role in translation of the 13 mtDNA encoded polypeptides which are all structural subunits of the respiratory chain complexes. Mutations in RMND1 lead to development of a mitochondrial cytopathy characterized by lactic acidosis, deafness, renal dysfunction and myopathy. Renal dysfunction is a relatively uncommon feature of mitochondrial disease suggesting mutations in RMD1 may preferentially affect kidney function. Presently there are no detailed published descriptions of the autopsy findings associated with RMND1 related mitochondrial cytopathy. Methods: Herein, we report the autopsy findings in a 4-year-old boy with mitochondrial cytopathy caused by a pathogenic mutation in RMND1. Full clinical and biochemical features of the index case were recently published. Briefly, he was born at term following a pregnancy complicated by oligohydramnios. Mild sensorineural hearing loss was detected at birth through routine newborn screening. At 2 months of age, he presented with failure to thrive, diarrhea and epileptic encephalopathy. His unusual clinical renal presentation included lactic acidosis and renal dysfunction (declining glomerular filtration rate, hypertension, hyperkalemia and hyponatremia). Psychomotor development was delayed with severe hypotonia and myopathic features. Skeletal muscle complexes I and IV were significantly reduced. Magnetic resonance imaging (MRI) revealed increased T2 signals throughout the white matter and delayed myelination. His clinical condition deteriorated and he expired primarily due to progressive chronic renal failure at 4 years of age. Results: At autopsy, renal abnormalities were noted including renal hypoplasia, diffuse glomerulosclerosis, tubular atrophy, calcification, interstitial fibrosis and inflammation. The liver was enlarged secondary to steatosis. Histochemical analysis of skeletal muscle showed diffuse reduction in cytochrome C oxidase activity, abnormal NADH, SDH and PAS staining, increase in lipid and mild type-2 fiber atrophy. Non-specific chronic pancreatitis and adrenal cortical lipid depletion were also present. The spleen showed unusual subintimal deposits in the arterioles associated with the periarteriolar lymphoid tissue; this histological finding was not seen elsewhere. Ultrastructural analysis of skeletal muscle and kidney demonstrated rectilinear electron dense mitochondrial inclusions. Conclusion: This case report is the first detailed description of anatomical abnormalities associated with the RMND1 mitochondrial cytopathic phenotype, which is of value to recognizing RMND1 related mitochondrial cytopathy at autopsy and further characterizing the pathophysiology of this abnormality.Biography
Email: almadani.n@gmail.com