| Research Article |
Open Access |
|
| Selvadurai Muralidharan, Janaki Sankarachari Krishnan Nagarajan*, Sachin Singh and Anil Dubala |
| Department of Pharmaceutical Analysis, J.S.S College of Pharmacy, Tamilnadu, India |
| *Corresponding author: |
Janaki Sankarachari Krishnan Nagarajan
Center for Advanced Drug Research and Testing
J.S.S.College of Pharmacy
Tamilnadu, India
Tel: +91-423-2443393
Fax: +91-423-2442937
E-mail: nagasaki2001@rediffmail.com |
|
| Â |
| Received October 05, 2012; Published October 26, 2012 |
| Â |
| Citation: Muralidharan S, Krishnan Nagarajan JS, Singh S, Anil D (2012) Bioequivalence Study of Simvastatin. 1:438. doi:10.4172/scientificreports.438 |
| Â |
| Copyright: © 2012 Muralidharan S, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. |
| Â |
| Abstract |
| Â |
| A simple, rapid and selective method was developed for estimation of simvastatin from human plasma. The method involves simple protein precipitation techniques using etofylline as internal standard. Chromatographic separation was carried out on a reversed phase C18 column using mixture of methanol: 2 mM ammonium acetate and 500 μl of 0.5% formic acid (80:20, v/v) at a flow rate of 1.0 ml/min with UV-VIS detection at 418.35 nm. The retention time of simvastatin and internal standard were 5.41 and 1.086 min, respectively. The method was validated and found to be linear in the range of 1.0-10.0 ng/mL. An open, randomized, two-treatment, two period, single dose crossover, bioequivalence study in 12 fasting, healthy, male, volunteers was conducted. After dosing, serial blood samples were collected for the period of 24.0 h. Various pharmacokinetic parameters including AUC0–t, AUC0–∞, Cmax, Tmax, T1/2, and elimination rate constant (Kel) were determined from plasma concentration of both formulations. Log transformed values were compared by analysis of variance (ANOVA) followed by classical 90% confidence interval for Cmax, AUC0–t and AUC0–∞ and was found to be within the range. These results indicated that the analytical method was linear, precise and accurate. Test and reference formulation were found to be bioequivalent. |
| Â |
| Keywords |
| Â |
| Pharmacokinetic studies; Validation; HPLC; Statistical analysis; Simvastatin |
| Â |
| Introduction |
| Â |
| Simvastatin is a hypolipidemic drug belonging to the class of pharmaceuticals called statins. It is used to control hypercholesterolemia (elevated cholesterol levels) and to prevent cardiovascular disease (Wikepedia et al.,). Simvastatin is a powerful lipid- lowering drug that can decrease low density lipoprotein (LDL) levels by up to 50%. It is used in doses of 5 mg up to 80 mg. Higher doses (160 mg) have been found to be too toxic, while giving only minimal benefit in terms of lipid lowering. There is no real effect on HDL and triglyceride levels. From recent research it has become apparent that simvastatin and other statins inhibit the progression of atherosclerosis beyond their effects on LDL. Many explanations have been proposed, for example it’s inhibitory affect on macrophages in the atherosclerotic plaque lesions. In one non-randomized study, simvastatin halved the risk of developing dementia or Parkinson's disease [1]. Limited analytical methods have been developed for the determination of simvastatin in biological samples along with high-performance liquid chromatography methods [1-7]. Therefore, the aim of the present investigation was to develop a new, sensitive HPLC method for the estimation of simvastatin in human plasma. The method was applied to a bioequivalence study of simvastatin 40 mg tablets. The outcome of a study depends upon the reliability, reproducibility and sensitivity of the analytical methodology employed. Therefore, the bioanalytical method was validated in accordance with USFDA guidelines prior to the initiation of the study. |
| Â |
| Experiment |
| Â |
| Materials |
| Â |
| Working standard of Simvastatin with 99.96% purity was obtained from German Remedies Ltd., Mumbai, India. Etofylline (purity 99.56%) working standard was obtained from Cadila Health Care Ltd., Ahmedabad, India. Acetonitrile (HPLC grade), obtained from Qualigens Fine Chemicals, Mumbai and potassium dihydrogen ortho phosphate, ortho phosphoric acid, methanol, and trichloroacetic acid (all analytical grade reagent) were purchased from S.D. Fine Chem. Ltd., Mumbai. In house mill Q water was used throughout the study. Fresh frozen human plasma used in the method development was obtained from the Vijay Hospital, Ooty, and was stored at −20 ± 2ºC until required. |
| Â |
| Instrumentation and chromatography |
| Â |
| The HPLC system consisted of a LC-MS 2010 A (Shimadzu Ltd., Japan), Auto injector port with 10 μl loop (Rheodyne, USA) and UVVIS detector (Shimadzu Ltd., Japan). The wavelength of the detector was set at 418.95 nm. Detector output was quantified on Lab Solution chromatography software. Separation was carried out on a Princeton SHER C18, 4.6 mm×100 mm, Japan, using mixture of methanol: 2 mM ammonium acetate and 500 μl of 0.5% formic acid (80:20, v/v) as a mobile phase, at a flow rate of 1 ml/min. Total analysis time was 15 min. All analysis was performed at oven temperature 30°C. |
| Â |
| Preparation of calibration standard |
| Â |
| Stock solutions of simvastatin and olanzapine (I.S) (1 mg/ml) were prepared in mixture of water and acetonitrile (1:1 V/V) and stored at 8°C. The stock solution of simvastatin was further diluted with the mixture of water and acetonitrile to give series of standard solutions. Calibration standard of simvastatin (10, 20, 40, 60, 80, 100 ng/ml) were prepared by spiking appropriate amount of the standard solution in blank plasma. |
| Â |
| Quality control standards |
| Â |
| Lowest quality control standards (LQC), median quality control standards (MQC) & highest quality control standards (HQC) were prepared by spiking drug free plasma with simvastatin to give solutions containing 2.0, 6.0, and 8.0 ng/ml, respectively. They were stored at −20 ± 2° C till analysed. |
| Â |
| Sample preparation |
| Â |
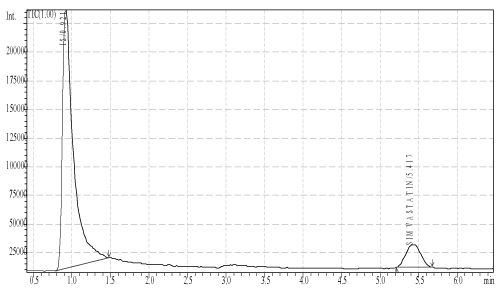
| To 0.5 ml plasma sample containing simvastatin (calibration standard), 0.5 ml of internal standard (10.0 μg/ml) was added and followed by 0.5 ml of 10% trichloroacetic acid was vortexed followed by centrifugation at 4000 rpm for 15 min at 4°C. The supernatant solution was separated and injected. None of the drug free plasma samples studied in this assay yield endogenous interference at these retention times Figure 1. |
| Â |
|
|
Figure 1: Typical chromatogram of sample solution. |
|
| Â |
| Validation |
| Â |
| The selectivity of the method was checked for interference from plasma. The standard curve consisting of five points ranging from 1.0 to 10.0 ng/ml was developed. Quality control samples i.e. LQC (2.0 ng/ml), MQC (6.0 ng/ml) and HQC (10.0 ng/ml) were used to determine the intra and inter-day precision and accuracy of the assay. Peak area ratio of simvastatin to internal standard were fit to linear equation (y=0.0012x-2E-06) and drug concentration in control samples along with the same day standard curve samples were calculated using this equation. For all the curves the correlation coefficients (r2) were never lower than 0.9982 [8]. |
| Â |
| Clinical design |
| Â |
| The study protocol was approved by The Inst itution Ethics Committee. Twelve healthy adult, male, human Indian subjects with mean age group 22.67 ± 2.27 years and average weight 61.67 ± 6.89 kg were included in the study. Subjects were excluded from the study if one of more of following criteria were present at time of medical screening: allergic to simvastatin, history or clinical data of renal or liver disease, positive test for hepatitis B, HIV, history of alcohol, drug addiction or donated blood within 72 days prior to study. Test and reference formulation of simvastatin 40.0 mg tablet were administered with 240 ml of water. The study was conducted according to the principles outlined in the declaration of Helsinki. The study was conducted as 12×2 single dose, randomized, open, and complete crossover design. Volunteers were fasted overnight before and 3 h after drug administration. Blood sample (5 mL) were collected at 0.00 h and 0.5,1.0,1.5,2.0,2.5,3.0,3.5,4.0,4.5,6.0,8.0,12.0 and 24.0 of post drug administration through an indwelling cannula into heparinised glass vials. After drug administration standard breakfast and lunch were provided at 3, 6, 9 and 12 h of post dose. The blood samples were immediately centrifuged, plasma was separated and stored at −20 ± 2°C until analysed. After a washout period of 7 days, the study was repeated in the same manner to complete the crossover design. The plasma samples obtained at various time intervals were analysed by the HPLC method developed. |
| Â |
| Pharmacokinetic analysis |
| Â |
| The plasma concentration profile obtained was fed into PK solution, computer software on Microsoft excel®, to determine the pharmacokinetic parameters. The maximum simvastatin concentration Cmax and the corresponding peak time Tmax were determined by the inspection of the individual drug plasma concentration–time profiles. The elimination rate constant Kel was obtained from the least-square fitted terminal log-linear portion of the plasma concentration–time profile. The elimination half-life T1/2 was calculated as 0.693/Kel. The area under the curve to the last measurable concentration (AUC0–t) was calculated by the linear trapezoidal rule. The area under the curve extrapolated to infinity (AUC0–∞) was calculated as AUC0–t+Ct/Kel where Ct is the last measurable concentration. |
| Â |
| Statistical analysis |
| Â |
| For the purpose of bioequivalence analysis AUC0–t, AUC0–∞ and Cmax were considered as primary variables. Bioequivalence of two formulations was assessed by means of an analysis of variance (ANOVA) for crossover design and calculating 90% confidence interval of the ratio of test/reference using log transformed data. The formulation was considered bioequivalent when the difference between two compared parameters was found statistically insignificant (p>0.05) and confidence interval for these parameters fell within 80–125% [9]. |
| Â |
| Results and Discussion |
| Â |
| Selectivity |
| Â |
| Selectivity of the method described was investigated by screening six different batches of human blank plasma. Under the proposed assay condition simvastatin and internal standard had a retention time of 5.056 and 1.086 min, respectively, rest of the peaks were due to the plasma components. Simvastatin and internal standard were very well resolved under the proposed chromatographic conditions. |
| Â |
| Accuracy |
| Â |
| The mean percent accuracy of the proposed method was found to be 97.93%. |
| Â |
| Precision |
| Â |
| Intraday precision for simvastatin was 1.887 ± 0.0721, 5.809 ± 0.1402, 9.851 ± 0.1256 for the spiked concentration at 2.0, 6.0 and 10.0 ng/ml and the percent coefficient of variation (%CV) was 3.82, 2.41 and 1.28, respectively. Inter day precision for simvastatin was 1.8466 ± 0.1063, 5.6940 ± 0.1863 and 9.5362 ± 0.2723 for the spiked concentration at 2.0, 6.0 and 10.0 ng/ml and the percent coefficient of variation (%CV) was 5.75, 3.27 and 2.86 respectively (Table 1). |
| Â |
|
|
Table 1: Precision studies. |
|
| Â |
| Linearity |
| Â |
| The linearity of each calibration curve was determined by plotting the peak area ratio of simvastatin to internal standard verses nominal concentration of simvastatin. For linearity study seven different concentration of simvastatin were analysed (1.0, 2.0, 4.0, 6.0, 8.0, 10.0 ng/ml). The peak area response was linear over the concentration range studied. Each experiment at all concentration was repeated three times on three separate days to obtain the calibration data. The coefficient of correlation ‘r2’ was found to be 0.9982. The limit of quantification and limit of detection were 5.0 and 1.0 ng/ml, respectively. |
| Â |
| Recovery |
| Â |
| The mean extraction recoveries of simvastatin determined over the concentration of 2.0, 6.0 and 10.0 ng/mL were 98.35%, 97.35% and 986.09%. For the internal standard (200 ng/mL), the mean extraction recovery was 97.93% (Table 2). |
| Â |
|
|
Table 2: Recovery Studies. |
|
| Â |
| Stability study |
| Â |
| Short-term and long-term stock solution stability study was evaluated, which proved no significant deviation from normal value when stored at 4°C. The stability of simvastatin in plasma was determined by measuring concentration change in quality control samples over time. Stability was tested by subjecting the quality controls to three freeze-thaw cycles and compared with freshly prepared quality control samples. As shown in (Table 3), the mean concentration of simvastatin in quality control samples did not change significantly within the time period under the indicated storage conditions. Long-term stability studies results conclude that simvastatin is stable in plasma matrix at least for 30 days when stored at −20 ± 2°C. |
| |
| Â |
|
|
Table 3: Stability of drug in plasma during storage and plasma handling. |
|
| Â |
| System suitability |
| Â |
| System suitability test was performed daily before the run of analytical batch to check detector response to the analyte. This method showed a good ruggedness, in fact little change in mobile phase ratio or normal laboratory condition of humidity, light, and air exposure temperature did not influence the retention time of simvastatin and internal standard. Both the formulations were well tolerated by all the volunteers in both the phases of study. No clinical adverse events occurred during the study. All calculated pharmacokinetic parameter values were in good agreement with the previously reported values. For bioequivalence evaluation, Cmax, AUC0–t, and AUC0–∞ were considered as primary parameters. The mean and standard deviation of these parameters of the two formulations were found to be very close, indicating that the plasma profiles generated by test formulation is comparable to those produce by reference formulation. Analysis of variance (ANOVA), after log transformation of the data, showed no statistically significant (p>0.05) difference between the two formulations. The mean peak plasma concentrations for 40.0 mg simvastatin tablet were found to be 70.0128 and 81.3045 ng/ml for the reference and test formulations respectively. AUC0–24 was found to be 56.8765 and 67.1555 ng.h/ml for the reference and test formulations respectively. AUC0–∞ was found to be 70.0128 and 81.3045 ng.h/ml for the reference and test formulations respectively. The elimination rate constant Kel for the reference and test formulation was found to be 0.0899 and 0.0899, respectively. |
| Â |
| Conclusion |
| Â |
| An HPLC–UV based method has been developed for quantification of simvastatin in human plasma. The sensitivity and simplicity of the method makes it suitable for pharmacokinetic studies. The statistical comparison of AUC and Cmax clearly indicated no significant difference in the two formulations of 40.0 mg of simvastatin tablets. Ninety percent confidence interval for the mean (T/R) of AUC0–t, AUC0–∞ and Cmax indicates that the reported values were entirely within the bioequivalence acceptance range of 80-125% (using log transformed data). Based on these results a concise decision on bioequivalence was taken. Hence it was concluded that formulation ‘Test’ is bioequivalent with formulation Reference. |
| Â |
| |
| References |
| Â |
- Wolozin B, Wang SW, Li NC, Lee A, Lee A, et al. (2007). Simvastatin is associated with a reduced incidence of dementia and Parkinson's disease. BMC Medicine 5: 20.
- Barrett B, Huclova J, Borek-Dohalsky V, Nemec B, Jelinek I (2006) Validated HPLC–MS/MS method for simultaneous determination of simvastatin and simvastatin hydroxy acid in human plasma. J Pharm Biomed Anal 41: 517-526.
- Carlucci G, Mazzeo P, Biordi L, Bologna M (1991) Simultaneous determination of simvastatin and its hydroxy acid form in human plasma by high-performance liquid chromatography with UV detection. J Pharm Biomed Anal 10: 693-697.
- Ali H, Nazzal S (2009) Development and validation of a reversed-phase HPLC method for the simultaneous analysis of simvastatin and tocotrienols in combined dosage forms. J Pharm Biomed Anal 49: 950-956.
- Novakova L, Satinsky D, Solich P (2008) HPLC methods for the determination of simvastatin and atorvastatin TrAC Trends in Analytical Chemistry 27: 352-367.
- Hefnawy M, Al-Omar M, Julkhuf S (2009) Rapid and sensitive simultaneous determination of ezetimibe and simvastatin from their combination drug products by monolithic silica high-performance liquid chromatographic column. J Pharm Biomed Anal 50: 527-534.
- Vuletic M, Cindric M, Koruznjak JD (2005) Identification of unknown impurities in simvastatin substance and tablets by liquid chromatography/tandem mass spectrometry. J Pharm Biomed Anal 37: 715-721.
- Guidance for Industry, Bioanalytical Method Validation (2001) Rockville, Maryland: U.S. Department of Health and Human Services. Food and Drug Administration, Center for Drug Evaluation and Research, CDER.
- Bolton S, Charles B (2009) Experimental Design in Clinical trials. In: Pharmaceutical StatisticsPractical and Clinical Applications(5th edn), New York: Marcel Dekker Inc. 384-443.
|
| Â |
| Â |