| Research Article |
Open Access |
|
| Michael Abrams, David Barton, Eamon Vandaei, Diana Romero, Adam Caldwell and Cleber Ouverney* |
| San Jose State University, Department of Biological Sciences, San Jose, CA 95192-0100 |
| *Corresponding authors: |
Cleber Ouverney
San Jose State University
Department of Biological Sciences
San Jose, CA 95192-0100
E-mail: cleber.ouverney@sjsu.edu |
|
| |
| Received June 30, 2012; Published August 28, 2012 |
| |
| Citation: Abrams M, Barton D, Ouverney C (2012) Genomic Characteristics of an Environmental Microbial Community Harboring a Novel Human Uncultured TM7 Bacterium Associated with Oral Diseases. 1:276. doi:10.4172/scientificreports.276 |
| |
| Copyright: © 2012 Abrams M, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. |
| |
| Abstract |
| |
| The diversity of prokaryotes associated with humans has been dominated by uncultured species (not isolated in pure culture). For instance, nearly 80% of the human gut and 68% of the human oral microbes are thought to be uncultured; some of which have been associated with human oral, digestive, vaginal, and cardiovascular diseases. The prevalence of uncultured pathogens is expected to continue to increase within the near future. In fact, public databases such as GenBank have nearly quadrupled the number of candidate phyla (those made entirely of uncultured organisms) since the 1980s and currently lists many new lineages of unclassified Bacteria and Archaea. Because detection of uncultured organisms has been limited to the 16S rDNA gene sequence, little is known about the function (e.g. pathogenicity) they play in humans. Access to human samples, however, imposes limitations to reproducible data and alternative models have been proposed. The work here presented attempts to further characterize the uncultured bacteria in the TM7 Candidate Division using both cultured-independent molecular and cultivation methods. Our long-term goal is to investigate how environmental strains of TM7 can serve as potential model organisms to help us determine the role uncultured bacteria play in humans. |
| |
| Keywords |
| |
| Uncultured bacteria; TM7 candidate division; Human pathogen; Fosmid library; 16S rDNA gene |
| |
| Introduction |
| |
| It has been estimated that the vast majority of cells associated with the human body are actually not human cells, but rather uncultured microbial cells. Uncultured microbes are those not yet isolated in pure culture. In fact, that same estimation claims that 90% of all cells that make up the entire human system are of prokaryotic origin. The human body has, on average, ten trillion (1013) human cells and attached to the human body are one hundred trillion (1014) prokaryotic cells. Hence, the human body is said to be only 10% human cells [1]. While the vast majority of prokaryotes are found in the human gut [2] the human oral flora (~108 cells/g of plaque) [3] and the skin (~103 cell/cm2) [4] are also colonized by a large number of microorganisms. Up to 80% of the human gut and 68% of the human oral microbes are thought to be uncultured. |
| |
| Equally intriguing is the diversity of microbial life found in association with humans. There are approximately 500 to 1,000 species of bacteria in the gut [2] or the skin [4], whereas the oral cavity harbors approximately 500-700 different species [5] of bacteria based on a combination of classical cultivation-based and culture-independent molecular methods. |
| |
| This large abundance and diversity become important to OMICS studies because each of the different types of microbial life contributes with a unique set of genes and proteins, which in turn, interact with the human body cells and can affect human health. It has been known that the establishments of a healthy microbial community help the human host better defend against invading pathogens. For many years, the molecular approaches applied to characterize microbial communities relied primarily on the 16S rDNA gene. Today, high-throughput next generation sequencing has revolutionized the field and genomic sequences have dominated the study of microbial diversity. |
| |
| Microbial abundance and diversity have also been observed in environmental habitats. Like in humans, the great majority of microbes in environmental samples lack representation in pure cultures. In seawater, for instance, 99.9% of the bacteria are thought to be uncultured [6]. Today, half of the ~52 phyla in the Bacteria Domain are made entirely of uncultured members. A phylum with no cultured representative is known as a Candidate Division. A particularly interesting Candidate Division is the TM7, which members have been detected in a diversity of environmental habitats [7], but also in association with the human skin [4], the human mouth [5], gut, stomach [1] and vaginal fluid. TM7a bacteria, a subgroup of the TM7, have been associated with gingivitis [8] and periodontitis [3,9], two human oral diseases that affect a large fraction of the population worldwide [10]. |
| |
| More recently, a homolog of oral TM7a was discovered in an environmental habitat sharing >99% identity in their 16S rDNA sequences [11]. It was our goal to further characterize the microbial community in that same environmental site where the TM7a-like bacterium was discovered using a combination of next-generation sequencing, 16S rDNA sequence analysis, and cultivation tools. |
| |
| Materials and Methods |
| |
| We used culture-independent methods to characterize an environmental habitat with a mixed and complex microbial community. A prior study at this same sampling site had identified a TM7 bacterium closely related to a TM7 homolog associated with human periodontal disease (a.k.a. TM7a) based on 16S rDNA gene sequence analysis [11]. In this investigation we used a genomic approach by generating a fosmid library with DNA extracted from that entire microbial consortium. Presence of TM7 in our sample was confirmed via cloning and sequence analysis of the TM7 16S rDNA gene. Finally, we attempted to isolate the TM7 bacterium from the study site and acquire the first pure culture belonging to the TM7 Candidate Division. |
| |
| Sample Collection and DNA Isolation |
| |
| Samples were collected from an activated wastewater tank in the San Jose/Santa Clara Water Pollution Control Plant (Santa Clara, CA) in sterile vials and kept in ice while transported to the lab for processing. This is the same tank from where the original TM7a homolog was discovered [11]. Once in the lab, the samples were well mixed by vortexing and aliquoted in 50ml volumes in sterile conical tubes. To generate the fosmid library, 50 ml of sample was centrifuged for 10 minutes at 3500 g and 4°C. One gram of the resulting pellet was used for genomic DNA extraction using the Meta-G-Nome™ DNA Isolation Kit (Epicentre, Madison, WI, catalog # MGN0910), which isolates approximately 40 kb unbiased DNA fragments. The protocol was followed according to the manufacturer's recommendations. |
| |
| DNA quality and concentration were assessed by a Nanodrop ND- 1000 spectrophotometer (Nanodrop Technologies™, Wilmington, DE). A 5 μl aliquot of the fragmented DNA was run on a 20 cm 0.8% agarose gel electrophoresis at 30 volts for 16 hours at room temperature, to confirm size, and then concentratedμl to 500 ng for direct use in the library construction. The presence of TM7 in the DNA extraction to be used was determined using PCR with primers BAC-8F (5’AGA GTT TGA TCM TGG CTC AG3’) and TM7-1177R (5’GAC CTG ACA TCA TCC CCT CCT TCC3’), which target the TM7 Candidate Division 16S rRNA gene [11]. |
| |
| Construction of fosmid and 16S rDNA gene libraries |
| |
| Samples testing positive for TM7 in the PCR above were used to construct a fosmid library using the CopyControl™ Fosmid Library Production Kit (Epicentre, Madison, WI, and catalog # CCFOS110) following the manufacturer's instructions. Randomly selected clones were digested with NotI restriction enzyme in order to estimate insert size and analyzed using agarose gel electrophoresis as described above. The fosmid library is a PCR-independent method, whereas the 16S rDNA gene library used a 1.2kb DNA fragment insert PCR amplified from the sample genomic DNA extraction. |
| |
| In parallel, a 16S rDNA gene library was constructed following the procedures detailed in Dinis et al. (2011). While the PCR above detected TM7 16S rDNA genes, the 16S rDNA clone library and sequencing, confirmed the presence of the TM7a phylotype in our samples. In brief, we cloned a 1,200 bp PCR amplicon of the 16S rDNA gene using Invitrogen pCR2.1-TOPO vector, and transformed into One Shot TOP10 competent cells, following Invitrogen protocols for bluewhite screening. |
| |
| Fosmid library PCR screening and analysis |
| |
| We also screened the Fosmid clones for the presence of 16S rDNA genes by pooling ~40 fosmids at a time. Each fosmid pool was induced for high-copy number overnight and fosmid DNA was extracted using the QIAprep™ Spin Miniprep Kit (Qiagen, Valencia, CA). Pooled fosmid DNA samples were screened for TM7 16S rDNA with primer pair BAC-8F and TM7-1177R [11]. Reaction conditions consisted of an initial denaturation step of 95°C for 2 min, and 40 cycles of 95°C for 30 sec denaturation, 61°C for 30 sec annealing, 72°C for 30 sec extension, and a final extension at 72°C for 5 min. |
| |
| Additionally, the broad-range primer pair DG74 forward (5AGG AGG TGA TCC AAC CGC A3') and 143 reverse (5'GAY GAC GTC AAR TCM TCA TGC3') was used to identify Gram-positive bacteria 16S rDNA gene in the sample and avoid amplification of the fosmid host bacterium E. coli 16S rDNA [12]. The PCR reaction conditions for the latter primers were previously described [12]. PCR products of correct size were extracted with an E-gel apparatus (Invitrogen, Carlsbad, CA), cleaned using Qiagen PCR cleaning kit and sequenced from both sides using the respective PCR primers. Sequence homology to known 16S rDNA was assessed using BLASTn and the non-redundant database [13]. |
| |
| Fosmid and 16S rDNA libraries sequencing |
| |
| Individual fosmid clones were induced in overnight culture and subsequently extracted using the QIAprep™ Spin Miniprep Kit (Qiagen, Valencia, CA, catalog #27106). Fosmid DNA was sequenced with vector primers T7-BACR3, or M13F-R. Sequences were cleaned and trimmed using CodonCode Aligner v3.7.1 (Dedham, MA). High quality sequences were uploaded onto the MG-RAST server for analysis. We also used the BLASTx and BLASTn non-redundant databases to determine the homology of our sequences to previously published sequences. |
| |
| For the 16S rDNA gene library, we first screened 300 white colonies for the correct insert size using vector M13F and M13R primers. Then we purified plasmids from 200 clones with QIAprep Spin Miniprep Kit (Qiagen, Valencia, CA) and submitted all 200 to be sequenced with M13F primer by Sequetech, Mountain View, CA. After sequence analysis (see below), clone sequences of interest were sequenced from both sides and ~1,200 bp contigs were used to generate a phylogenetic tree. |
| |
| TM7 cultivation on filter membrane |
| |
| Cultivation of TM7 from the sludge sample was done on the surface of a 0μm.2 isopore polycarbonate membrane filter as previously described [8]. |
| |
| The PC filter containing a highly diluted (10-4 to 10-6) bacterium inoculum was placed over a sterile tissue culture insert (TCI) (Nunc, Roskilde, Denmark) with cells facing up (Figure 1A). Nutrient to support bacterial cell growth was supplied by filling the TCI with a nonsterile solution made of neat sludge sample from where the TM7 bacteria were detected originally. The TCI was made with μm a rigid 0.02 and hydrophilic filter, which allowed free diffusion of macromolecules and dissolved nutrients from the sludge medium to the bacterial cells on top of the PC filter, while preventing large particles and bacterial cells from contaminating the cultures. |
| |
|
|
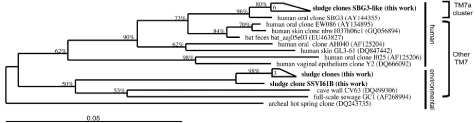
Figure 1: Phylogeny of TM7 16S rDNA gene sequences. The tree used 20 TM7 16S rDNA gene sequences from both environmental and human sources. The TM7a cluster included phylotypes from environmental habitat in this study (sludge), which were ≥ 99.0% homologous to the humanassociated oral counterparts. The tree was built using ARB software package and Archaea was used as the outgroup. |
|
| |
| Each of 16 TCI devices was kept inside a well of a tissue culture plate and incubated in a clean surface at room temperature for several weeks. Once per week cells on the PC filter were resuspended in sterile 1X PBS and a small volume of the resuspension was screened via PCR for TM7 using the BAC-8F and TM7-1177R primers as described above. TM7- positive cultures based on PCR were prepared for fluorescence in situ hybridization (FISH) as described below. The remaining resuspended cells in 1X PBS were transferred to a sterile PC filter for further growth in clean TCI devices. |
| |
| FISH of TM7 microcolony |
| |
| Fluorescence in situ hybridization was performed on 50% ethanolfixed bacterial cells growing on the surface of the PC filters to validate PCR results. FISH also provided crucial information about the TM7 cell morphologies as well as abundance in the culture. FISH technique used TM7 specific probe TM7-905 (5’CCG TCA ATT CCT TTA TGT TTT A3’) labeled with a Cy3 fluorochrome as previously described [3]. In addition to the probe, bacterial cells were labeled with the general DNA-binding dye YOPRO-1 (Invitrogen, Carlsbad, CA), which labeled all cells containing DNA in the sample. FISH-labeled whole cells were visualized using a Zeiss Confocal Laser Scanning microscope model LSM-510 and FISH images were captured using the Zeiss image capture software. |
| |
| Results |
| |
| 16S rDNA gene sequence analysis |
| |
| 16S rDNA clone library confirmed the presence of TM7a in our samples (Figure 1). Out of the 200 clones sequenced from the 16S rDNA gene library, 120 sequences (60%) belonged to the TM7 Candidate Division and 84 of those clones (~70%)≥98had.5% similarity to the human TM7a group [11]. Sequences≥ sharing 98.5% similarity in 16S rDNA gene means the sequences belong to the same OTU (operational taxonomic unit), which can be loosely regarded as the same species of bacteria. We established the evolutionary relationships among 10 of our TM7 sequences against 10 publicly available TM7 reference sequences from human and environmental sources (Figure 1). An archaeal sequence was used as outgroup to root the tree. Six of our sludge 16S rDNA sequences clustered with the human oral clone SBG3, which belongs to the TM7a group [11]. The other four 16S rDNA from our sludge samples clustered with other environmental TM7. |
| |
| Fosmid library PCR screening |
| |
| |
| The metagenomic library analyzed 4012 individually resolved clones, and based on the titer, an additional 4000 clones remain unresolved in long term storage. Twenty randomly selected clones digested with NotI restriction enzyme yielded an average insert size of 35 kb. PCR screening using primer set BAC-8F and TM7- 1177R detected one uncultured Betaproteobacteria clone with a 98% BLASTn similarity to accession number CU922341, amplified from an anaerobic sludge digester in France. The DG74 and143 PCR primer pair detected one uncultured Actinobacteria (98% similar to GenBank accession GQ487893) amplified from a soil polluted by heavy metals (unpublished) in China and one uncultured Clostridia (98% similar to DQ011249) detected in a stratum sewage from an oil field, also in China (unpublished). |
| |
| Fosmid sequence analysis |
| |
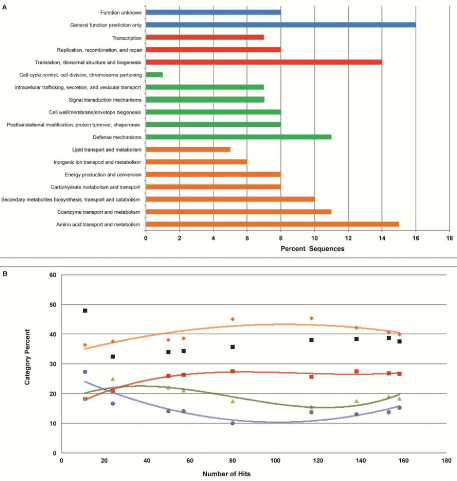
| A total of 421 fosmid end sequences, with average length of 707 bp and an average GC content of 61%, were compared to the Cluster of Orthologous Groups (COG) database using MG-RAST (Figure 2). The analysis generated 158 hits. The remaining 263 end-sequences had no hit in COG categories, suggesting the presence of novel genes and untapped genomes. Shown in Figure 2A, carbohydrate, amino acid, and lipid metabolism and transport were identified as the most abundant category with 39.87% of the hits. Categories “cellular processes” and “signaling” constituted 26.58% of the reads, and the subcategory “defense mechanisms” was the most abundant. The COG category “information storage” and “processing” was responsible for 18.35% of the hits, while the “poorly characterized” category made up 15.19% of the hits. |
| |
|
|
Figure 2: Fosmid end sequences compared to the Cluster of Orthologous Groups (COG) database.
(A) Major COG categories consisted of color-coded sub categories: (Blue) Poorly Characterized, (Red) Information Storage and Processing, (Green) Cellular Processes and Signaling, and (Orange) Metabolism. Parameters for sequence comparison were set using the recommended limits of 1e-5 e-Value cutoff, a minimum identity of 60 percent, and a minimum alignment length of 15 base pairs.
(B) Change in percent of each major COG category as fosmid sequence hits increased. At each point, the sequences were classified using the same COG categories in figure 2A. As an increasing number of clones were sequenced, each category approaches saturation. The black data points represent the percentage of hits relative to the number of sequences for each respective x coordinate. |
|
| |
| COG distributions over the number of sequence hits are plotted in Figure 2B to demonstrate a stabilization of data as more sequences are analyzed. BLASTx searches of the sequences also detected an end sequence with a best hit to an extracellular solute binding protein pertaining to the uncultured candidate division OP1 bacterium, providing evidence that a genomic fragment of OP1 may have been recovered within our fosmid library. Thus far, no genome has been sequenced from the OP1 cluster of the Bacteria Domain. |
| |
| GenBank searches through MG-RAST detected 9 genes that code for antibiotic resistance proteins, 5 of which were acriflavin resistance genes. One of the acriflavin hits had a 97% amino acid similarity to the acriflavin resistant protein derived from Delsufovibrio magenticus, and another hit had 88% amino acid similarity to Candidatus Accumulibacter phosphatis clade IIA, indicating that these bacteria may be present in this sludge environment. Acriflavin genes such as AcrB are well characterized in E.coli, and are prevalent in Gram-negative bacteria (9). Due to the abundance of acriflavin-like resistance genes, it is likely that there is significant environmental pressure caused by pollution. Previously studies have shown that antibiotic-contaminated environments exhibit resistance genes at frequencies as high as 1.7% [14]. Our sequencing efforts here have determined an antibiotic resistance gene frequency of 2.14%. |
| |
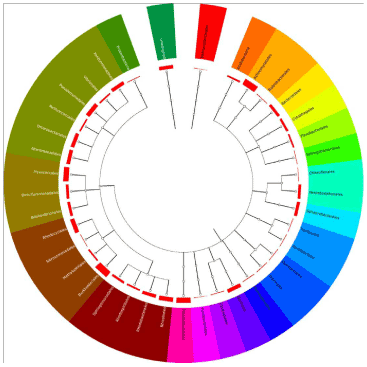
| The end sequences were also compared to the SEED database through MG-RAST for phylogenetic analysis (Figure 3). Of the 221 hits produced, 214 clustered within the Bacteria Domain, whereas one clustered within the Archaea Domain. The remaining six sequences were unable to be clearly assigned to a category. The most abundant phylum in our sample based on this SEED analysis was Proteobacteria, with 62.2% of the hits, followed by Actinobacteria (14.95%), Planctomycetes (6.07%), and Bacteroidetes (4.67%). Chloroflexi, a sister group to the Candidate Division TM7, had 10 hits representing 4.67% of the total hits. All other phyla were under 4% of the total hits. Betaproteobacteria, particularly the order Burkholderia, was prevalent, making up 76.92% of the class. In the Gammaproteobacteria, the order Pseudomonadales was most abundant with 27.27% of the class. |
| |
|
|
Figure 3: Phylogeny of fosmid sequences. Fosmid end sequences were uploaded onto MG-RAST for phylogenetic analysis, and were compared to the SEED database. The standard conditions of 1e-5 e-Value cutoff, a minimum identity of 60 percent, and a minimum alignment length of 15 base pairs were used. Each branch point represents an order, and each bar is logarithmically proportional to the relative abundance. |
|
| |
| TM7 microcolony and FISH |
| |
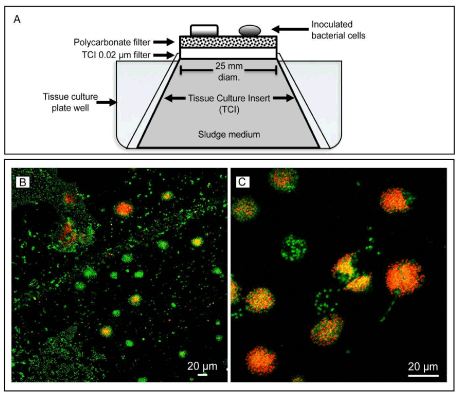
| Cultivation confirmed the presence of several TM7 FISH-labeled microcolonies (Figure 4B and 4C). The TM7 microcolonies were detected within 2 weeks of cultivation and ranged in size from 15 to 30μm in diameter and were commonly formed by a discrete cluster made of several hundred cells. Exact quantification was difficult due to cell overlap. TM7 single-cell sizeμmaverageandthe1 most common morphologies were cocci, diplococci, and short bacilli. All microcolonies labeled with the TM7 FISH probe also showed numerous unlabeled cells, indicating a mixture of cells present in each microcolony. |
| |
|
|
Figure 4: TM7 cultivation set up.
(A) Diluted sludge sample containing bacterial cells was inoculated on top of a 0.2 μm polycarbonate (PC) filter membrane sitting over the 0.02 μm filter of the Tissue Culture Insert (TCI). Nonsterile sludge medium held inside the TCI diffused to the bacterial cells. The entire apparatus was kept inside a tissue culture well.
(B) And (C) Micrographs of 2-week old culture showing bacterial cells labeled with a TM7 specific probe (orange-red) using FISH technique. Cells formed microcolonies on the PC filter. YOPRO-1, a DNA-binding dye, was used to label all cells (green). Notice that the TM7 microcolonies (orange-red) are mixed with other non-TM7 cells (green). Scale bars = 20 μm. |
|
| |
| Discussion |
| |
| Fosmid-based metagenomics allows for the efficient cloning of large genomic fragments, as well as confident gene prediction. Here, we used a moderate number of fosmid end sequencing coupled with 16S rDNA PCR screening to elucidate nuances of a known uncultured bacterium’s environment. Based on COG analysis, diverse metabolic genes were abundant, thus implying diverse potential of the microbial community. The low percentage of fosmid ends that were classified (37.5%) would be expected in an environment as varied as sludge, and most likely represents limitations of the COG database or the presence of novel genes with no known homology. The sequences analyzed in this study have strikingly similar COG hits and distribution when compared to deep-sea sediment fosmid libraries, indicating that smaller data sets can act as representative models [15,16]. A large percentage of antibiotic resistance genes were discovered, indicating that the microbial community in the sample site was forced to adapt to a wide range of selective pressures [11]. |
| |
| The majority of sequences were classified as Proteobacteria and Actinobacteria, while only residual viral and Archaea sequences were detected. Additionally, a number of fosmid ends were classified as uncultured bacteria, particularly the Candidate Division OP1 and Candidatus Accumulibacter phosphatis clade IIA. |
| |
| The 16S rDNA library and phylogeny confirmed the presence of TM7, including TM7a in our sampling site. Furthermore, TM7 microcolonies validated the viability of the TM7 cells in our samples. Microcolonies, however, were composed of a mixture of cells, suggesting TM7 requires co-cultivation in order to grow. Also, whether TM7 and the other (non-TM7) nearby cells maintain a symbiotic relationship (mutualistic or commensal) is yet to be determined. Despite not being able to isolate a pure culture of TM7, our results were exciting and we plan to use micro manipulation such as microfluidics [17] or flow cytometer [18] to further separate the TM7 microcolonies and use next generation sequencing approach to determine the bacterial cell types. OMICS studies can potentially ais in determining the genetic makeup and metabolic potential of TM7 as well as its requirement for growth and eventually lead us to isolate this unique and novel group in pure culture. |
| |
| Future goals are to acquire complete fosmid sequences for the identification of gene structure and operons, providing useful data for accurate taxonomic binning and functional classification. Full sequencing of all fosmids inserts in the library will allow for more indepth analysis and characterization of this waste water environment, providing insight into the functional pathways and microbial species that interact with the Candidate Division TM7. Our long-term goal is to better understand the role TM7 bacteria play in human health. However, difficulties to access human samples on a continuous basis have, in part, hindered an efficient characterization of this uncultured novel bacterium. We propose that an environmental homolog, such as TM7a-like in our environmental site, could serve as a model organism to expedite our understanding of the TM7 potential pathogenicity. |
| |
| |
| References |
| |
- Bik EM,Eckburg PB,Gill SR,Nelson KE,Purdom EA, et al. (2006) Molecular analysis of the bacterial microbiota in the human stomach. Proc Natl Acad Sci U S A 103: 732-737.
- Gill SR, Pop M, Deboy RT, Eckburg PB, Turnbaugh PJ, et al. (2006) Metagenomic analysis of the human distal gut microbiome. Science 312: 1355-1359.
- Brinig MM, Lepp PW, Ouverney CC, Armitage GC, Relman DA (2003) Prevalence of bacteria of division TM7 in human subgingival plaque and their association with disease. Appl Environ Microbiol 69: 1687-1694.
- Grice EA, Segre JA (2011) The skin microbiome. Nat Rev Microbiol 9: 244-253.
- Dewhirst FE, Chen T, Izard J, Paster BJ, Tanner AC, et al. (2010) The human oral microbiome. J Bacteriol 192: 5002-5017.
- Jannasch HW, Jones GE (1959) Bacterial populations in sea water as determined by different methods of enumeration. Limnology and Oceanography 4: 128-139.
- Hugenholtz P, Tyson GW, Webb RI, Wagner AM, Blackall LL (2001) Investigation of candidate division TM7, a recently recognized major lineage of the domain Bacteria with no known pure-culture representatives. Appl Environ Microbiol 67: 411-419.
- Ferrari BC, Binnerup SJ, Gillings M (2005) Microcolony cultivation on a soil substrate membrane system selects for previously uncultured soil bacteria. Appl Environ Microbiol 71: 8714-8720.
- Kumar PS, Griffen AL, Barton JA, Paster BJ, Moeschberger ML, et al. (2003) New bacterial species associated with chronic periodontitis. J Dent Res 82: 338-344
- Armitage GC (1999) Development of a Classification System for Periodontal Diseases and Conditions. Ann Periodontol 4: 1-6.
- Dinis JM, Barton DE, Ghadiri J, Surendar D, Reddy K, et al. (2011) In search of an uncultured human-associated TM7 bacterium in the environment. PLoS One 6: e21280.
- Klausegger A,Hell M,Berger A,Zinober K,Baier S, et al. (1999) Gram type-specific broad-range PCR amplification for rapid detection of 62 pathogenic bacteria. J Clin Microbiol 37: 464-466.
- McGinnis S, Madden TL (2004) BLAST: at the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res 32: W20-25.
- Kristiansson E, Fick J, Janzon A, Grabic R, Rutgersson C, et al. (2011) Pyrosequencing of antibiotic-contaminated river sediments reveals high levels of resistance and gene transfer elements. PLoS One 6: e17038.
- Hu Y, Fu C, Yin Y, Cheng G, Lei F, et al. (2010) Construction and preliminary analysis of a deep-sea sediment metagenomic fosmid library from Qiongdongnan Basin, South China Sea. Mar Biotechnol (NY) 12: 719-727.
- Martín-Cuadrado AB, López-García P, Alba JC, Moreira D, Monticelli L, et al. (2007) Metagenomics of the deep Mediterranean, a warm bathypelagic habitat. PLoS One 2: e914.
- Marcy Y, Ouverney C, Bik EM, Lösekann T, Ivanova N, et al. (2007) Dissecting biological "dark matter" with single-cell genetic analysis of rare and uncultivated TM7 microbes from the human mouth. Proc Natl Acad Sci U S A 104: 11889-11894.
- Kalyuzhnaya MG, Zabinsky R, Bowerman S, Baker DR, Lidstrom ME, et al. (2006) Fluorescence in situ hybridization-flow cytometry-cell sorting-based method for separation and enrichment of type I and type II methanotroph populations. Appl Environ Microbiol 72: 4293-4301.
|
| |
| |