| Research Article |
Open Access |
|
| Vivekananda Sarangi and Somnath Tagore* |
| Dept. of Biotechnology & Bioinformatics, Dr DY Patil University, Plot 50, Sec 15, CBD Belapur, Navi Mumbai – 400614, India |
| *Corresponding authors: |
Somnath Tagore
Dept. of Biotechnology & Bioinformatics
Dr DY Patil University, Plot 50, Sec 15, CBD Belapur
Navi Mumbai–400614, India
E-mail: somnathtagore@yahoo.co.in |
|
| |
| Received February 03, 2012; Published August 24, 2012 |
| |
| Citation: Sarangi V, Tagore S (2012) Cost Analysis: Its Role in Metabolomics. 1: 246. doi:10.4172/scientificreports.246 |
| |
| Copyright: © 2012 Sarangi V, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. |
| |
| Abstract |
| |
| A significant path in a metabolic pathway can be helpful in finding out the evolutionary relationship of pathways, can be used to produce suitable vectors for commercial purpose and also be used for drug designing. Though shortest path is mostly the significant one but other ways of finding significance can give different results. The other ways include assigning cost or weights to the metabolites and the enzymes. Then depending on the criteria on which the weights are assigned, the significant path is found out. Cost can be assigned based on the degree of a particular metabolite; it can also be the probability value of a metabolite being converted to another. Negative value of the degree of a metabolite can also be assigned as weights. Depth first searching and graph partitioning is used for finding all possible paths and LPP is used for finding out the significant one based on more than one condition. Here we have done a comprehensive review of the current analysis and techniques in this area and have proposed a hypothesis for assigning costs. |
| |
| Keywords |
| |
| Cost analysis; Depth first search; Graph; Path enumeration; Significant path |
| |
| Introduction |
| |
| Metabolic pathways consist of series of chemical reactions taking place inside the living cell. Most of the pathways in the body are connected to each other and are important for the maintenance of homeostasis within an organism. Basically in a pathway a particular metabolite is converted into another by an enzyme [1]. These are the components of a pathway and are denoted by nodes and edges in a quantitative manner using graphs. Sometimes the nodes denote the enzyme and edge denote the metabolites and other times the reverse. In this paper we will be referring to nodes as metabolites and edges as enzymes [2]. |
| |
| The quantitative approach of analysing consists of assigning weights to the components of the pathway. Each node or each edge has a natural weight in the pathway (.i.e. how important the particular component is) [2]. This weight is given externally to the components and based on these weights calculations are performed. These calculations may include finding out a significant path or finding out the how important a particular edge (enzyme) or node (metabolite) is in the transformation of metabolite ‘X’ to metabolite ‘Y’ [3]. |
| |
| The basic necessity is to find out the significant path. It can give us the best path for the maximum production of a desired metabolite. For large scale production of human enzymes in microorganism, this significant path can be incorporated in the vector, which is then introduced in the microorganism colony [4]. It saves a lot of time and energy which is otherwise wasted in hit and trial method. Furthermore by studying diseased pathways we can find out one or few intermediate steps which are present in all possible paths. So finding a way to remove these steps from the cell can cause the diseased process to stop. Before going to find the significant path it is important to find out all the possible paths between the two metabolites [5]. Then we are in the position to find out what makes the significant path more significant than other paths. If a particular portion of the path is present in all the paths and it constitutes the significant path, it can be said that this short portion is conserved over evolution. Hence finding significant path and comparing it with all possible paths can throw light on the evolution of pathway [6]. |
| |
| In order to assign weights to the nodes and edges and then to find the significant path we first need to convert the pathway into a graph form. Thus the metabolites are treated as nodes and the enzymes are treated as edges. It is represented as G (V, E). Where V= {v1, v2, ...., vn} corresponds to the set of vertices/metabolites and E={e1, e3 ,....en} corresponds to the set of edges/enzymes [7]. The manuscript is divided into several sections like introduction (already discussed), techniques involved and analysis strategies, proposed methodology, current scenario, discussion and reference. |
| |
| Techniques Involved and Analysis Strategies |
| |
| One can store the reactants and products in separate files and then using them as vertex to create edges between them, thus forming the entire graph. This is a tedious process and can lead to human errors [7]. But it is the easiest way once one has collected the entire required dataset. Another way of doing it is by using ‘sbml’ file of the pathway. It contains all the graphical data (edges and nodes) of a pathway [8]. |
| |
| Once the pathway is stored in graphical form the next hurdle is to find out all the possible paths between any two given metabolites. One way to do so is to find out the total number of vertices in the graph and then find out all the possible paths that are present between the given metabolites assuming it is a totally connected graph [9]. This is followed by using the adjacency matrix where the edges that are actually present are retained and the rest are discarded. Though not an impossible task, but finding out all possible paths in a totally connected graph, can be very intensive for the processor. A more feasible method of doing so is by using a combination of Depth First Searching (DFS) and Graph Partitioning Method (GP) [10]. After we have all the possible paths our next goal is to find the most significant one. For this purpose we need to do calculations on the paths, and for that we need to give weight to each component; assigning weights to the graph can be done in a number of ways depending on the type of result one wants like connectivity of vertices, edge relaxation to name a few [11]. |
| |
| Once the weight is assigned the next step is to find the most significant path from all the possible paths. Normally the shortest path is the most significant one (this is not always true). For finding the shortest path between two metabolites many algorithms are present such as Dijkstra's algorithm solving the single-pair, singlesource, and single-destination shortest path problems; Floyd-Warshall algorithm solving all pair’s shortest paths; k shortest path algorithm which finds the k shortest paths between a start and an end vertex in a graph [12]. Another way of finding significant path is by using Linear Programming Problem (LPP). A significant path is found out based on a single cost analysis strategy. But when we try to combine two or most cost analysis strategies together, it is not possible for a single linear equation to handle it. We have used a method called simplex method which employs LPP for solving many linear equations together [13]. |
| |
| Proposed Methodology |
| |
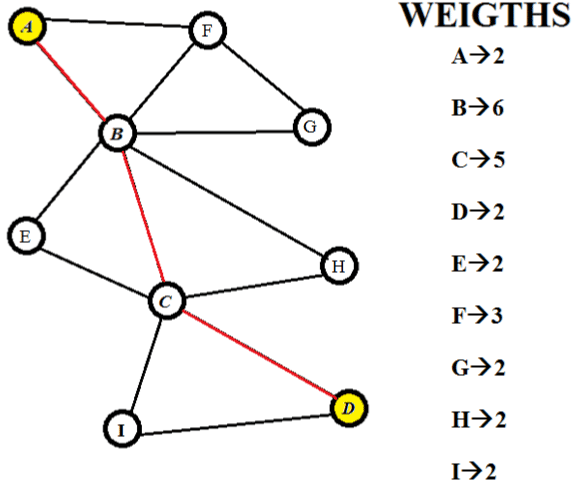
| Let us consider a hypothetical pathway. It has nine nodes and thirteen edges. The nodes are A, B, C, D, E, F, G, and H. We will be assigning weights to the nodes only. There are various methods of assigning costs/weights to nodes. The weights are assigned on the basis of connectivity of vertices. So the cost of each vertex is equal to the number of edges it has. (Figure 1) shows the entire pathway with the cost of each node on its right side [14]. |
| |
|
|
Figure 1: Hypothetical pathway and its corresponding weights. |
|
| |
| Now let us take two metabolites as start and end point. Let ‘A’ be the Start metabolite and ‘D’ the end metabolite. So first we have to generate all the possible paths. There are in total 18 paths possible. Each path is given a score by adding the score of the nodes present in it [15]. Then the scores will be compared and the path with the least score is the most significant one. |
| |
| All the paths are enumerated and the scores have been calculated (Figure 2). After comparing it is found that the path with the least score is A-B-C-D. Thus this is the most significant. Now when we compare all the paths between ‘A’ and ‘D’ we find that the metabolite ‘B’ and ‘D’ are present in all the paths. And only these two metabolites are present in the significant path [16]. This shows that these two metabolites have to be present for the conversion of ‘A’ to ‘D’ and for this reason they are very important as there is no alternate way without them [2]. It can also be proposed that this particular pathway is conserved over evolution and other paths were developed over a period of time when and where the necessities arouse [17]. |
| |
| |
|
|
Figure 2: Enumerated paths and their corresponding scores. |
|
| |
| Current Scenario |
| |
| Pathway reconstruction for finding drugs against malaria parasite Plasmodium falciparum has being carried out in Bioinformatics Center, Institute for Chemical Research, Kyoto University, Kyoto, Japan. The lack of malaria vaccine and resistance of the parasite to drugs were the main motivators. It is focused on the KEGG metabolic pathway of the parasite [18]. In the pathway there are many gaps in the annotations, which make the design of drugs difficult. Thus in order to bridge the gap virtual enzymes (Vzymes) are used along with KEGG orthologs clusters to resolve the missing enzymes. The enzymes thus found can be used as a drug target to combat the disease [18]. |
| |
| The distance between pathways is defined as the distance between the substrate in them and thus the distance between the corresponding enzymes. Thus it can show evolutionary relationship. It combines sequence information with information underlying the reaction network. The distance between the metabolites show the distance between the gene coding it. The change in this distance is showed by mutation. Hence studying the distance phylogenetic analysis of the pathways is performed [19]. Minimal pathway distance is the minimum number of metabolic steps separating two enzymes in a pathway. To do so the minimum distance was found out using linear programming. E.coli small molecule metabolism was used to find out the relation between the distance of two metabolites and its correlation with its genetic distance. |
| |
| Some of the tool in the field is described next. Path finding approaches in metabolic pathways have shown a sudden acceleration after 2003. PathMiner is another tool released in 2006 which predicts metabolic routes by reasoning over transformations using chemical and biological information. It is used in computer based Diagnostics [20]. Another tool called PHT (Pathway Hunter Tool) released in 2005 finds the k-shortest path between metabolites in a pathway [21]. ReTrace is a computational method for inferring branching pathways in genome-scale metabolic networks. It has a local copy of KEGG LIGAND database in directory kegg; one is able to compute branching pathways from metabolite X to metabolite Y. It is python based [22]. Another tool called MetaRoute released in 2008 is used for finding relevant metabolic routes for interactive network navigation and visualization [23]. |
| |
| Discussion |
| |
| Study of pathways can be used as an answer to many problems such as evolutionary relations, drug designing, better understanding of the biological systems, and large scale production of bio molecules such as enzymes. Most of the pathways are documented in case of higher organism like human. The most predictable use of cost analysis strategies for finding significant path is in the field of disease diagnosis, especially in the case of untreatable diseases as their course of action is not recognized, or because of the unknown changes it does in a particular pathway which leads to a series of faulty functions e.g. cancer. Better study of pathway can increase the scope of treatment. Comparison of healthy and diseased pathway can exactly tell where the fault lies. Furthermore diseased pathways may also show an abnormal significant path in comparison to normal which is facilitating the production of toxic metabolite in the patient. There are many ways where we can rely on this strategy, but like any other field it also has drawbacks. In a pathway a particular metabolic path is favoured over others. Its nature’s way of assigning weights; this is not well understood. So when we assign weights to the pathway artificially it is not exactly the way it is in nature. Mostly it is far from perfection. Thus we have to hypothesize our result with the limited number of data we get as output. There are chances of mistakes. This is the reason we cannot apply it directly to any field. Whatever the theory is it has to undergo a lot of tests. But the positive side of this method is that it reduces the time of the tester by narrowing down the search for tests. Therefore, makes the progress faster and easier. |
| |
| |
| References |
| |
- Beasley JE, Planes FJ (2007) Recovering metabolic pathways via optimization. Bioinformatics 23: 92-98.
- Klamt S, Gagneur J, Von Kamp A (2005) Algorithmic approaches for computing elementary modes in large biochemical reaction networks. Syst Biol (Stevenage) 152: 249-255.
- Wright J, Wagner A (2008) The Systems Biology Research Tool: evolvable open-source software. BMC Syst Biol 2: 55.
- Forst CV, Schulten K (2001) Phylogenetic analysis of metabolic pathways. J Mol Evol 52: 471-489.
- Klamt S, von Kamp A (2009) Computing paths and cycles in biological interaction graphs. BMC Bioinformatics 10: 181.
- Srikant R, Sundaram R, Singh KS, Pandu Rangan C (1993) Optimal path cover problem on block graphs and bipartite permutation graphs. Theoretical Computer Science 115: 351-57.
- Jeong, H, Tombor B, Albert R, Oltvai ZN, Barabási AL (2000) The large-scale organization of metabolic networks. Nature 407: 651-654.
- Kanehisa M, Goto S, Kawashima S, Nakaya A (2002) The KEGG databases at GenomeNet. Nucleic Acids Res 1: 42-46.
- Deo N (1974) Graph Theory with application to Engineering and Computer science. Prentice-Hall.
- Kim BJ, Yoon CN, Han SK, Jeong H (2002) Path finding strategies in scale-free networks. Phys Rev E Stat Nonlin Soft Matter Phys 65: 27103.
- Kingsford CL, Chazelle B, Singh M (2005) Solving and analyzing side-chain positioning problems using linear and integer programming. Bioinformatics 21: 1028-1036.
- Allman ES, Rhodes JA (2004) Mathematical models in biology. Cambridge University Press.
- Serrano MA, Boguñá M, Vespignani A (2009) Extracting the multiscale backbone of complex weighted networks. Proc Natl Acad Sci USA 106: 6483-6488.
- Sitharam M (2004) Counting and enumeration of self-assembly pathways for symmetric macromolecular structures. Nova Southeastern University, Fort Lauderdale, Florida, USA, Advances in Bioinformatics and its Applications December 16-19, 2004.
- Klamt S, Haus UU, Theis F (2009) Hypergraphs and Cellular Networks, PLoS Comput Biol 5: e1000385.
- Pitkänen E, Rantanen A, Rousu J, Ukkonen E (2005) Finding Feasible Pathways in Metabolic Networks. In: Lecture Notes in Computer Science Berlin / Heidelberg Springer 123-133.
- Kanehisa M, Limviphuvadh V, Okuno Y, Katayama T, Goto S, et al. (2003) Metabolic Pathway Reconstruction for Malaria Parasite Plasmodium falciparum. Genome Informatics 14: 368-369.
- Simeonidis E, Riso SC, Thornton J.M, Bogle, ID and Papageorgiou LG (2003) Analysis of metabolic networks using a pathway distance metric through linear programming. Metab Eng 5: 211-219.
- McShan DC, Rao S, Shah I (2003) PathMiner: predicting metabolic pathways by heuristic search. Bioinformatics 19: 1692-1698.
- Rahman SA, Advani P, Schunk R, Schrader R Schomburg D (2005) Metabolic pathway analysis web service (Pathway Hunter Tool at CUBIC). Bioinformatics 21: 1189-1193.
- Pitkänen E, Jouhten P, Rousu J (2009) Inferring branching pathways in genome-scale metabolic networks, BMC Syst Biol 3: 103.
- Blum T, Kohlbacher O (2008) MetaRoute: fast search for relevant metabolic routes for interactive network navigation and visualization. Bioinformatics 24: 2108-2109.
|
| |
| |