| Research Article |
Open Access |
|
| Dennis G. Karounos1,2,4,5,6*, Jason A. Brandon5, Terry Lacy2, J. Scott Bryson3,5 |
| 1Medical Service, VAMC, University of Kentucky, Lexington, KY USA |
| 2Division of Endocrinology, and Molecular Medicine, University of Kentucky, Lexington, KY USA |
| 3Division of Hematology and Blood & Marrow Transplantation, Departments of Internal Medicine, University of Kentucky, Lexington, KY USA |
| 4Physiology, University of Kentucky, Lexington, KY USA |
| 5Microbiology, Immunology, & Molecular Genetics, University of Kentucky, Lexington, KY USA |
| 6Center for Nutritional Sciences, University of Kentucky, Lexington, KY USA |
| *Corresponding authors: |
Dennis G. Karounos, M.D
Chief, Endocrinology Section VA Medical Center
1101 Veterans Drive Rm. B402
Lexington, KY 40502
E-mail: dkaroun@uky.edu |
|
| |
| Received December 01, 2011; Published July 27, 2012 |
| |
| Citation: Karounos DG, Brandon JA, Lacy T, Bryson JS (2012) Role of Regulatory T Cells in the Prevention of Diabetes by Insulin in NOD Mice. 1: 161. doi:10.4172/scientificreports.161 |
| |
| Copyright: © 2012 Karounos DG, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. |
| |
| Abstract |
| |
| The use of insulin therapy to prevent development of type 1 diabetes in nondiabetic relatives of individuals with diabetes has been difficult. We demonstrate that in animal models, it was necessary to use high dose insulin to prevent diabetes. To mimic the conditions of the Diabetes Prevention Trial, DPT-1, we administered insulin without adjuvants by daily subcutaneous injections to young adult prediabetic mice. To confirm that diabetes was prevented (rather than just delayed) treatment was continued for 70 to 80 weeks and glucoses were monitored weekly. We evaluated T cell function and phenotype using adoptive transfer, and regulatory T cell staining by flow cytometry. High dose insulin therapy provided significant protection from diabetes (diabetes free survival at age 72 wks of 56% versus 17 % survival in the untreated control group (p =0.00034,). The number of pathogenic T cells were reduced but not completed eliminated by long-term insulin preventative therapy. In contrast, there was a significant increase in CD4+FoxP3+ T cells with insulin therapy (7.6% + 0.4 SEM) compared to prediabetic mice (4.4% + 0.2) or overtly diabetic mice (3.6% + 0.2, P<0.0001). In untreated control mice, 15-20% of the mice did not develop disease. Analysis of these naturally-protected mice showed that they also had an increase in CD4+FoxP3+ Tregs. Comparison of Tregs in insulin-protected compared to naturally-protected mice demonstrated presence of insulin specific CD4+FoxP3+ Tregs in insulin treated mice but not in naturally protected animals. At the initiation of insulin-preventative therapy, if mice were given anti-GITR antibody DTA-1, the insulin preventative therapy was less effective and diabetesonset was accelerated. |
| |
| Our results demonstrate that an animal model of spontaneous diabetes, longterm therapy with high dose insulin prevents diabetes, and that induction of a regulatory subset of lymphocytes may be an important factor to prevent the development of type 1 diabetes. |
| |
| Keywords |
| |
| Diabetes mellitus; Insulin; Autoimmunity; Regulatory T cells; Immunetherapy |
| |
| Abbreviations |
| |
| NOD: Nonobese Diabetic Mice; Treg: Regulatory T cell; mAb: Monoclonal Antibody; wks: Weeks; LN: Lymph Node |
| |
| Introduction |
| |
| Type 1 diabetes mellitus is an immune-mediated metabolic disorder characterized by the destruction of the pancreatic beta cells by both humoral and cell-mediated immune mechanisms. This results in hyperglycemia and metabolic derangements requiring lifelong insulin therapy to sustain life. A hallmark of this chronic autoimmune disease is the lymphocytic infiltration of the islets that develops prior to the clinical onset of disease and is present both in man [1] as well as in animal models of diabetes [2]. Additional proof that this is an immune mediated disorder is the observation that treatment after clinical onset of diabetes with immunosuppressive agents can induce an insulinfree remission of the disease [3]. However, the toxicity associated with the use of immunosuppressive agents has limited this approached as an immune therapy of type 1 diabetes. This has resulted in increased interest in the use of more selective therapies such as the administration of autoantigens to induce protective immune responses and prevent the development of diabetes. Insulin is an important autoantigen in type 1 diabetes. Autoantibodies to insulin are detected prior to initiating insulin therapy [4] and are an important immunological marker of the disease [5]. |
| |
| The nonobese diabetic (NOD) mouse, an animal model of spontaneous diabetes, shares many features of human type 1 diabetes including the abrupt onset of overt diabetes, the dependence on exogenous insulin to sustain life, the presence of lymphocytic infiltration of the pancreatic islet cells before the onset of hyperglycemia, and the prevention of disease by immunotherapy. Insulin specific T cell clones have been isolated from NOD mice and have been shown to be capable of adoptively transferring diabetes in NOD mice [6]. A number of studies have demonstrated that insulin is capable of generating protective immune responses. In preclinical studies, subcutaneous administration of insulin to prediabetic animals has prevented or delayed the development of clinical disease [7,8]. Zekzer and colleagues generated an insulin-specific T cell clone from the pancreatic lymph node of an NOD mouse and demonstrated the ability of this clone to block the spontaneous development of diabetes in NOD mice [9]. However, even with the increasing amount of preclinical data, the translation of insulin-specific therapy for the prevention of diabetes has been difficult. In an NIH multicenter, randomized controlled clinical trial, Diabetes Prevention Trial, DPT-1, in persons at high risk for diabetes, low dose insulin therapy did not delay or prevent the development of type 1 diabetes [10,11]. |
| |
| In an effort to overcome challenges encountered in the clinical trials, we designed our preclinical studies in NOD mice to mimic the conditions of the clinical trials. We initiated therapy in young adult mice after the onset of insulitis using subcutaneous injections of insulin without adjuvants. We demonstrate that insulin preventative therapy protected young adult mice from diabetes even when initiated after the onset of extensive lymphocytic infiltration of the islets [8]. The purpose of this study is to determine the mechanism of this protection by insulin preventative therapy. We evaluated T cell function and phenotype using adoptive transfer, and regulatory T cell staining by by flow cytometry. Our data demonstrate that while the onset of clinical diabetes was prevented with insulin given to prediabetic mice, the lymphocytic infiltrates persisted and splenocytes from insulinprotected mice were still capable of transfering disease using an adoptive transfer model. However, with insulin therapy, or with mice naturally protected from the development of diabetes, there was an increase in CD4+FoxP3+ regulatory T cells (Tregs) in the lymphoid tissues of treated animals. Furthermore, in the insulin-protected mice there was an increase in the numbers of insulin-specific Tregs. Finally, in mice simultaneously treated with insulin therapy and anti-GITR mAb (DTA-1), an antibody shown to inhibit the activity of Tregs, the insulin protective response was blocked suggesting that Treg cells play a pivotal role in the prevention of diabetes by insulin. |
| |
| Materials and Methods |
| |
| Animals |
| |
| Female NOD/MrkTacfBR mice (Taconic Farms, Inc., Germantown, NY), or NOD/ShiLtJ mice and NOD.CB17-Prkdcscid/J (Jackson Laboratory Bar Harbor, ME) were purchased at 6-8 weeks of age and were housed under specific-pathogen-free conditions. A spontaneous incidence of diabetes of 64% at age 20 weeks, and 80% at age 36 weeks, with median onset of diabetes of 26 weeks (range 16-52 wks) was observed for the NOD/MrkTacfBR mice and a spontaneous incidence of diabetes of 30% at age 20 weeks, and 75% at age 36 weeks, with median onset of diabetes of 26 weeks (range: 12-59 wks) for NOD/ ShiLtJ mice. The animal use protocol was approved by the local Veterans Administration Institutional Animal Use and Care Committee. |
| |
| Treatment |
| |
| Female NOD mice, age 8 weeks, were randomly divided into treatment groups. Just prior to initating therapy, an additional two animals were sacrificed to confirm the degree of insulitis prior to initiating therapy. The animals received either insulin (human ultralente, Novo Nordisk, NJ), or human glargine insulin (Sanofi-Aventis, NJ) diluted with hormone-free suspension diluent fluid: 0.16% m-cresol, 0.065% phenol, 1.6% glycerol, 0.2% sodium phosphate and water, pH 7.4 for ultralente or pH 4.5 for glargine insulin) 0.1 ml subcutaneously each day. To mimic conditions of the DPT-1, we administered insulin to young adult prediabetic mice by daily subcutaneous injections, 5 days per week given at 5 microgram/day. The 5 microgram/day dose was derived by administering insulin at increasing levels and monitoring glucoses periodically after the injections. This was the maximum dose of insulin that could be givenwithout hypoglycemia. Mice were observed for hypoglycemia and if any hypoglycemia was observed, the dose of insulin was reduced to 2.5 micrograms/day for one to two weeks and then increased as tolerated. Control animals received hormone-free vehicle or no injections. |
| |
| All animals were evaluated for the onset of diabetes by monitoring blood glucose at one week intervals. Insulin was withheld for 48 hours to avoid masking diabetes onset by insulin therapy, and blood glucose was then monitored. In addition, the animals were monitored carefully when receiving their daily injections. If polyuria or weight loss was noted, blood glucose was evaluated. Any elevated blood glucose was verified by repeat analysis, with diabetes diagnosis confirmed when two consecutive blood glucoses were >16.7 mmol/L (300 mg/dl). Diabetesfree survival was calculated using the product limit or Kaplan-Meier method, and analysis of variance and T test calculated using GraphPad Prism software (San Diego, CA). |
| |
| Degree of Insulitis |
| |
| Randomly-selected animals from each group were sacrificed and the pancreas examined in a double-blind fashion for insulitis. Histological analysis of the pancreas was performed using the technique of Serreze et al. with minor modifications. Briefly, pancreas were fixed in formalin, stained with hematoxylin and eosin and sectioned at three nonoverlapping levels. Six to ten islets per mouse were individually scored by two independent observers using a semiquantitative scale ranging from 0 to 4: 0, normal islet with no sign of T cell infiltration; 1, islet associated with perivascular, peri-ductal leukocytic infiltration only; 2, more extensive peri-islet infiltration but with lymphocytes with less than 25% islet destruction; 3, >25% islet destruction; 4, complete islet destruction. An insulitis score for each mouse wasobtained by dividing the total score for each mouse by the number of islets examined. |
| |
| Adoptive Transfer |
| |
| Adult NOD.CB17-Prkdcscid/J mice were acclimated in our facility for at least 4 wks and used as secondary recipients. Lymphocytes (2x107/0.1 ml) were isolated from the spleens of NOD donor mice and were administered to NOD/scid hosts by intraperitoneal injection from diabetic, donors, or from insulin-protected donors. The NOD/ scid mice were 24 wks of age and the NOD/Lt diabetic donor mice were 24 wks of age with blood glucoses of 375 to 605 mg/dl on at least two occasions. The insulin-protected donors were 74 weeks of age and had received either glargine or ultralente insulin therapy for 62 wks by daily subcutaneous injection five days per week. Each week the insulin therapy was held for 48 hours and the glucose was monitor to determine if the mice had developed diabetes. After adoptive transfer, the mice were followed for development of diabetes by monitoring for blood glucose two times per week. |
| |
| Preparation of lymphoid cell suspensions |
| |
| Control and insulin-treated mice were euthanized by CO2 inhalation and lymphoid tissues removed (spleens, mesenteric lymph nodes (LN), pancreatic LN). Single cell suspensions were prepared from individual spleens and LN pooled within each group, by physical disruption of the tissue and the red blood cells were lysed by treatment with 0.83% Tris-buffered NH4Cl. The resulting cell population was counted using trypan blue exclusion. |
| |
| Detection of insulin-specific regulatory T cells |
| |
| Lymphoid cells were resuspended into 10% complete RPMI (10% FCS, 100 U/ml penicillin and 100 μg/ml streptomycin and 2 mM glutamine, 5x10-5 M of 2-ME). 2x106 cells were added in 1ml to wells of a 24-well tissue culture plate and were stimulated with 300 ng/ml of insulin (Roche, Indianapolis). Treated and untreated cells were cultured for 72 h at 370C in a CO2 incubator, then harvested and stained for expression of CD4 and intracellular FoxP3 as described. |
| |
| Flow cytometry analysis |
| |
| To monitor for the presence of CD4+FoxP3+ regulatory T cells 1x106 spleen cells from treated animals or following in vitro stimulation with insulin were placed in staining buffer (PBS containing 1% FCS, 0.1% NaN3) and incubated with anti-CD16/CD32 (Fc Block; BD PharMingen, San Diego, CA) to reduce non-specific surface staining. The cells were then stained with fluorochrome-conjugated mAb against CD4 (Caltag Burlingame CA). Intracellular staining for FoxP3 was performed using the Mouse Regulatory T cell Staining Kit (eBioscience, San Diego, CA) according to manufacturer’s directions. The cells were analyzed using a BD Biosciences FACSCalibur flow cytometer (San Jose, CA). |
| |
| Results |
| |
| Insulin protection from diabetes in young adult NOD mice with established insulitis |
| |
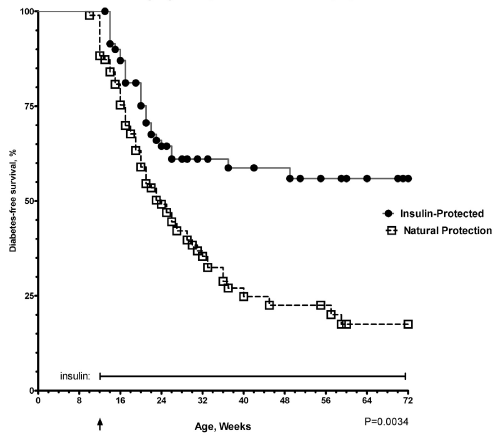
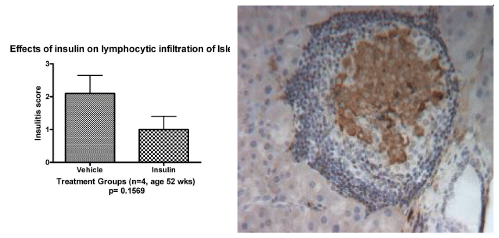
| To mimic the conditions of recent clinical trials [10], we gave daily subcutaneous injections of insulin without adjuvants (five days per week) to young adult prediabetic mice after they already developed severe insulitis [8]. We have now treated a larger cohort of mice with insulin compared to untreated control animals to give us the opportunity to compare immunological parameters in mice with insulin-protected diabetes compared to untreated mice that have natural protection of diabetes. Mice receiving insulin had significant protection from the development of diabetes compared to the age matched control mice with a diabetes-free survival of 56% compared to only 17% in the control group at 72 weeks of age after 60 weeks of insulin therapy (log rank Mantel-Cox Chi square 15.74, P= 0.0034, Figure 1) . .In mice protected from the development of diabetes with there appears to be lifelong protection from diabetes. In follow-up studies for greater than 72 weeks of age we rarely see any insulinprotected mice develop diabetes after the thirty-six week time point (Figure 1). However, evaluation of the pancreatic islets revealed that the lymphocytic infiltrates persisted over time (Figure 2a). Even though the islets were surrounded by periislet infiltrates, there was abundant staining for insulin (Figure 2b) and the blood glucoses remained in the normal range. |
| |
|
|
Figure 1: Insulin protection from diabetes in young adult NOD mice with established insulitis NOD/shjlt (black circles, n=72) mice were treated with daily subcutaneous injections of glargine insulin 5 mcg/day, five days per week. Blood glucoses were monitored weekly. In addition, NOD control mice (open squares, n=96) were monitored for development of diabetes to identify a cohort of mice that were naturally protected from development of diabetes. The larger number of mice for this study was to provide adequate numbers of mice for further characterizations of differences in naturally-protected mice, insulin protected mice and overtly diabetic mice. |
|
| |
|
|
Figure 2: Persistence of Lymphocytic Infiltration of the Islets in NOD mice Protected from Diabetes with Insulin Pancreatic islets were examined for lymphocytic infiltrates in NOD mice and insulitis score was determined in female NOD mice protected from development of diabetes by treatment for 52 weeks with daily subcutaneous injections of insulin compared to mice treated with insulin diluent (vehicle alone). There was significantly greater protection from diabetes in mice treated with insulin compare to mice receive vehicle alone. Even though mice were protected from development of diabetes, the lymphocytic infiltrates still persist (Figure 2a). The insulin treated mice tended to have fewer lymphocytic infiltrates compared to vehicle treated mice but the difference were not significantly different (P=0.1569). In mice protected from diabetes by insulin (age 74 wks) (Figure 2b), the lymphocytic infiltration persists, but there is abundant endogenous insulin staining and the mice maintain normal blood sugars. |
|
| |
| Lifelong insulin-preventative therapy does not eliminate diabetogenic effector T cells |
| |
| |
| To determine if this protection was due to elimination of diabetogenic effector cells, adoptive transfer experiments were perfomed transferring splenocytes from overtly diabetic or insulinprotected donors to NOD/scid recipients. Unexpectedly, splenic lymphocytes from insulin-treated mice were still able to transfer diabetes to NOD/scid hosts even though the donor mice were protected from diabetes for over 74 weeks (Figure 3). Splenocytes from overtly diabetic mouse donors transferred diabetes more rapidly than splenocytes from insulin-protected donors. With insulin-treated donors, diabetes onset was delayed with cells from the ultralente insulin donors (P=0.0228, logrank) and glargine-protected donors (P=0.0029, log-rank) compared to the more rapid transfer of diabetes using overtly diabetic donor mice (Figure 3). However, even though the donor mice had lifelong protection from diabetes, secondary recipients of splenocytes from the insulin-protected donors, still developed diabetes in 73-90% of the recipient animals. These results suggested that the protection from diabetes by insulin therapy was due to an active immune suppression and not insulin-mediated deletion of effector T cells. |
| |
|
|
Figure 3: Diabetogenic effector T cells persist in insulin-protected mice Using an adoptive transfer model, we evaluated whether diabetogenic cells were still present in mice insulin-protected mice. Donor splenocytes were obtained from overtly diabetic mice or from 74 wk old NOD mice protected from development of diabetes by 62 wks of daily ultralente or glargine insulin injections. The NOD.CB17-Prkdcscid/J recipients, age 24 wks, were injected with 2.0X107 purified donor splenocytes from ultralente insulin-protected mice (n=9, gray circles), glargine insulin-protected mice (n=11, gray triangles) or the overtly diabetic mice (n=9, black squares). The NOD/cb17 scid recipients were monitored for diabetes twice weekly. |
|
| |
| Increase in regulatory T cells with insulin preventative therapy |
| |
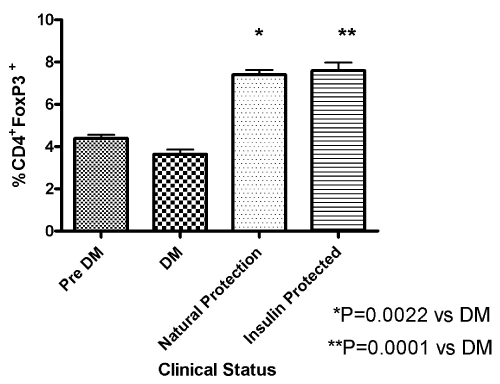
| The fork head protein Foxp3 has been identified as an important transcriptional regulator of Treg cells. To evaluate whether insulin therapy induced an increase in regulatory T cells, we examined splenocytes from insulin-protected mice (n=11) who remained free from diabetes after sixty weeks of treatment compared to animals with overt diabetes (n=7) or untreated prediabetic NOD mice (n=6). The incidence of diabetes in our colony was 75-80% at 35 wks of age for untreated mice so we would expect that at least 4-5 of these prediabetic mice will develop diabetes. Following intracellular staining for Foxp3 a significant increase in the regulatory T cell subset (CD4+Foxp3+) was observed in insulin protected mice that were treated for 60 weeks with insulin compared to the prediabetic or overtly diabetic mice (P= 0..0001, unpaired T test vs diabetic mice, Figure 4). In addition, a small percentage of NOD mice have natural protection and never develop diabetes. In these naturally-protected NOD, there was also a significant increase in CD4+Foxp3+ lymphocytes compared to the overtly diabetic mice (P=0.0022 unpaired T test, Figure 4). |
| |
|
|
Figure 4: Association of Treg with diabetes protection To evaluate whether insulin therapy induced an increase in regulatory T cells, we examined splenocytes from insulin-protected (n=11), animals with overt diabetes (n=7) or untreated prediabetic NOD mice (n=6), The spleen cells were stained for suface CD4 and intracellular FoxP3 as described. In these naturallyprotected NOD, there is also a significant increase in CD4+Foxp3+ lymphocytes compared to the overtly diabetic mice. |
|
| |
| Characterization of regulatory cells generated by insulinprotective therapy |
| |
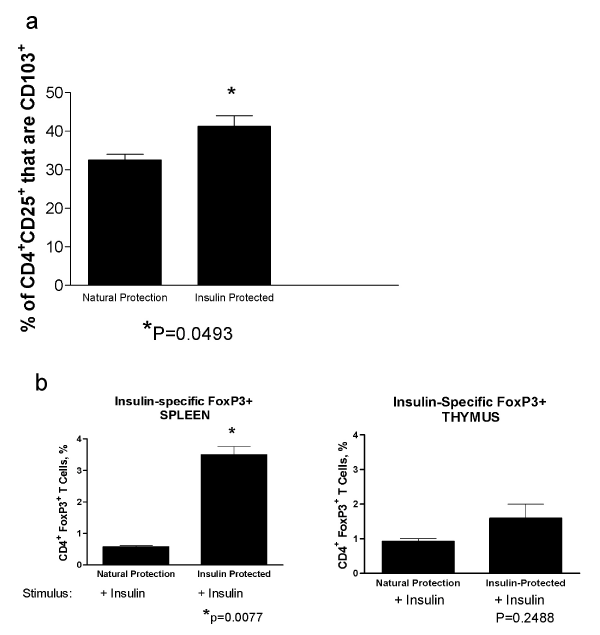
| Integrin CD103 (αEβ7) has been shown to be an important marker for identifying in vivo-activated CD4+FoxP3+ regulatory T cells [12]. We evaluated the presence of these CD4+CD25+CD103+ Treg by flow cytometry in spleen cells isolated from NOD mice (age 30 wks) who were protected from diabetes after 18 wks of insulin therapy compared to age-matched control mice that had natural protection from diabetes. There was a significant increase in the CD4+CD25+CD103+ Treg in the insulin treated mice (P=0.0493, T test, Figure 5a). |
| |
|
|
Figure 5: Generation of insulin-specific Tregs with insulin-preventative therapy A. Spleen cells isolated from NOD mice (age 30 wks) who were protected from diabetes after 18 wks of insulin therapy and age-matched control mice that had natural protection from diabetes. The presence of CD4+CD25+CD103+ Treg was determined by flow cytometry. There was a significant increase in the CD4+CD25+CD103+ Treg in the insulin treated mice (P=0.0493, T test). B. To determine if there is an increase in antigen-specific Treg associated with insulin therapy, splenocytes from insulin-protected and naturally-protected mice were incubated in vitro for 72 hrs with insulin and then analyzed for CD4 andFoxp3 expression by flow cytometry. There was a significant increase in the insulinspecific Foxp3 cells in the insulin-protected mice compared to mice that are naturally-protected from diabetes (P=0.0077, T test) in the spleen (left panel) but not in the thymus (right panel). |
|
| |
| To determine if there was an increase in antigen-specific Treg associated with insulin therapy, splenocytes from insulin-protected and naturally-protected mice were incubated in vitro for 72 hrs with insulin and then the Foxp3 expression determined by flow cytometry. There was a significant increase in the insulin-specific Foxp3 cells in the insulin-protected mice compared to mice that were naturallyprotected from diabetes in the spleen (Figure 5b, left panel, P=0.0077, T test) but not in the thymus (Figure 5b, right panel, ns, unpaired T test) Increases in Foxp3 and Treg-associated surface markers suggest that insulin protective therapy stimulated the development of antigenspecific Treg activity. |
| |
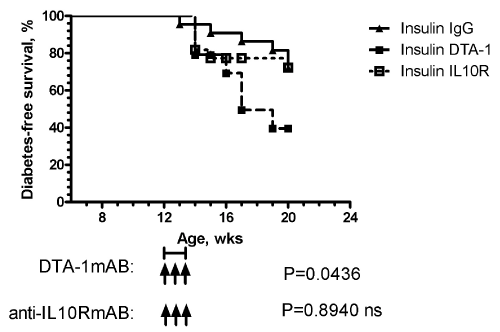
| To evaluate the functional importance of the insulin-induced regulatory Tcell response, NOD mice were simultaneously injected with insulin (5 days/week) and with the agonist monoclonal anti-GITR antibody DTA-1 weekly for three weeks beginning at 12 weeks of age in prediabetic female NOD mice. As a control, groups of animals were also treated with rat IgG. Administration of the anti-GITR antibody disrupts the ability of insulin to prevent the development of diabetes, while mice treated with insulin and rat IgG had significant protection from diabetes (Figure 6). In the DTA-1 treated mice, diabetes developed rapidly, occurring 4-6 week after antibody administration. |
| |
|
|
Figure 6: Anti-GITR antibody DTA-1 disrupts insulin mediated protection Simultaneous subcutaneous insulin therapy (five days per week) and the the agonist monoclonal anti-GITR antibody DTA-1 or rat IgG was administer weekly for three weeks by intraperitoneal injection beginning at 12 weeks of age. As in Figure 1, mice were monitored weekly for the development of diabetes. Administration of the anti-GITR antibody disrupted the ability of insulin to prevent the development of diabetes (log rank P=0.0436), while mice treated with insulin with rat IgG or insulin with anti-IL10R (log rank P=0.8940 compared to IgG) have significant protection from diabetes. |
|
| |
| Discussion |
| |
| The studies presented in the current manuscript extend and expand our previous work demonstrating the ability of insulin therapy to modulate the development of diabetes in the NOD mouse model system [8]. Results were presented that addressed the mechanisms by which insulin therapy alters the development of diabetes. Prevention of disease by insulin therapy was accompanied by increased production of insulin-specific regulatory T cells in the lymphoid tissues of treated animals. Inactivation of the regulatory cells by treatment with anti- GITR antibody resulted in enhanced diabetes, demonstrating the functional significance of the insulin-therapy-induced increased production of Treg. |
| |
| In unimmunized, normal adult mice, approximate 10% of peripheral CD4+ cells and less than 1% of CD8+ in normal adult mice express CD25+ [13]. Numerous studies document the important role of regulatory CD4+CD25+ T cells (Tregs) that express the transcription factor FoxP3 to maintain self-tolerance and control immune responses (reviewed [14]). Transgenic mice with the FoxP3 scurfy mutation and a diabetogenic T cell receptor BDC 2.5, have accelerated lymphocytic infiltration of their islets and dramatic progression to overt diabetes [15]. Thus, Tregs have an important role in protection from the development of diabetes. There appears to be two important subsets of these regulatory T cells including “natural” CD4+CD25+ from the thymus and “adaptive” CD4+CD25+ that are induce in the periphery derived from CD4+CD25- T cells if the appropriate antigenic stimulation and cytokine environment are present. Insulin therapy to prediabetic mice appears to induce this peripheral protective immune response. |
| |
| Our results demonstrate that daily subcutaneous insulin therapy started after the onset of severe insulitis can still prevent the onset of diabetes if given at high dose. Through our long-term follow-up, we demonstrate that diabetes onset is prevented and not just delayed. Lymphocytic infiltration of the islets persists even in mice protected from diabetes. Splenocytes from insulin-protected mice are still capable of transferring diabetes to NOD/scid hosts suggesting the presence of active immune suppression and not insulin-mediated deletion of effector T cells. However, long-term daily subcutaneous insulin therapy induces a protective regulatory response as demonstrated by the presence of CD4+FoxP3+ splenocytes in insulin-protected mice. Compared to mice that are naturally-protected from diabetes, there are equal numbers of CD4+FoxP3+ regulatory cells but development of diabetes is significantly reduced in mice with the insulin-induced regulatory cells (diabetes free survival of 17% vs 56%, respectively, P=0.0001). |
| |
| In the insulin-protected mice there was a significant increase in the insulin specific FoxP3+ T cells compared to mice that have natural protection from diabetes. Furthermore, these T cells are CD103+CD4+. In studies in graftversus-host disease the in vivo-activated CD103+CD4+ regulatory T cells are more effective at reducing inflammation than naturally-occurring Tregs [12]. Our data would suggest that these cells may also be important in insulin-protection from diabetes. |
| |
| The daily subcutaneous administration of antigen without adjuvant appears to provide an optimal environment to induce protective immune responses that are mediated by GITR+ regulatory cells. Our previous study [9] demonstrated that inactive insulin is equally effective at preventing type 1 diabetes as metabolically-active insulin. Thus, it is the immunological properties of insulin that are important for preventing type 1diabetes. The advantage of using an inactive insulin analog, is that it is possible to use higher doses of insulin without the risk of developing hypoglycemia in prediabetic individuals to induce this protective immune response and prevent type 1diabetes. |
| |
| Conclusions |
| |
| Our results demonstrate that in animal models of diabetes, longterm therapy with high dose insulin prevents diabetes, and induces an insulin specific peripheral regulatory T cell response. Induction of a regulatory subset of lymphocytes may be an important factor to prevent the development of type 1 diabetes. The fact that treated mice do not develop diabetes after long-term therapy provides evidence that clinical diabetes is prevented rather than simply being delayed. A key point about the model system presented herein is that we evaluated the ability of subcutaneous injections of insulin, without any adjuvants, to prevent spontaneous diabetes since this would have more relevance for translation into future clinical trials. Our data suggests that the prevention of diabetes by daily subcutaneous injection of insulin is mediated by the generation of insulin-specific regulatory cells. Thus, the subcutaneous delivery of antigen provides a favorable environment for induction of antigen-specific Treg and preventing the clinical onset of Type 1 diabetes.. In future clinical trials, monitoring the development of insulin-specific Treg may be a helpful parameter for improving efforts to use insulin to prevent the development of type 1 diabetes in man. |
| |
| Acknowledgements |
| |
| This project was supported in part by VA Merit Review Grant, Dept. of Veterans Affairs and the American Diabetes Association Research Grant. We thank Lee Ann Smith, Nicole Moore, and Betty Caywood for their expert technical assistance. |
| |
| |
| References |
| |
- Gepts W (1965) Pathologic anatomy of the pancreas in juvenile diabetes mellitus. Diabetes 14: 619-633.
- Rossini AA, Appel MC, Williams RM, Like AA (1977) Genetic influence of the streptozotocin-induced insulitis and hyperglycemia. Diabetes 26: 916-920.
- Stiller CR, Laupacis A, Dupre J, Jenner MR, Keown PA, et al. (1983) Cyclosporine for treatment of early type i diabetes: Preliminary results. N Engl J Med 308: 1226-1227.
- Palmer JP, Asplin CM, Clemons P, Lyen K, Tatpati O, et al. (1983) Insulin antibodies in insulin-dependent diabetics before insulin treatment. Science 222: 1337-1339.
- Sosenko JM, Krischer JP, Palmer JP, Mahon J, Cowie C, et al. (2008) A risk score for type 1 diabetes derived from autoantibody-positive participants in the diabetes prevention trial-type 1. Diabetes Care 31: 528-533.
- Daniel D, Gill RG, Schloot N, Wegmann D (1995) Epitope specificity, cytokine production profile and diabetogenic activity of insulin-specific T cell clones isolated from nod mice. Eur J Immunol 25: 1056-1062.
- Gotfredsen CF, Buschard K, Frandsen EK (1985) Reduction of diabetes incidence of bb wistar rats by early prophylactic insulin treatment of diabetes prone animals. Diabetologia 28: 933-935.
- Karounos DG, Bryson JS, Cohen DA (1997) Metabolically inactive insulin analog prevents type i diabetes in prediabetic nod mice. J Clin Invest 100: 1344-1348.
- Zekzer D, Wong FS, Wen L, Altieri M, Gurlo T, et al. (1997) Inhibition of diabetes by an insulin-reactive CD4 T-cell clone in the nonobese diabetic mouse. Diabetes 46: 1124-1132.
- Diabetes Prevention Trial--Type 1 Diabetes Study Group (2002) Effects of insulin in relatives of patients with type 1 diabetes mellitus. N Engl J Med 346: 1685-1691.
- Skyler JS, Krischer JP, Wolfsdorf J, Cowie C, Palmer JP, et al. (2005) Effects of oral insulin in relatives of patients with type 1 diabetes: The diabetes prevention trial-type 1. Diabetes Care 28: 1068-1076.
- Zhao D, Zhang C, Yi T, Lin CL, Todorov I, et al. (2008) In vivo-activated CD103+CD4+ regulatory t cells ameliorate ongoing chronic graft-versus-host disease. Blood 112: 2129-2138.
- Sakaguchi S, Sakaguchi N, Asano M, Itoh M and Toda M (1995) Immunologic self-tolerance maintained by activated t cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol 155: 1151-1164.
- Homann D, Von Herrath M (2004) Regulatory T cells and type 1 diabetes. Clin Immunol 112: 202-209.
- Chen Z, Herman AE, Matos M, Mathis D, Benoist C (2005) Where CD4+CD25+ Treg cells impinge on autoimmune diabetes. J Exp Med 202: 1387-1397.
|
| |
| |