| Research Article |
Open Access |
|
| Arash Fazeli1*, Nadali Babaeian Jelodar1, Farveh Ehya2, Laleh Krarimi Farsad2, Mostafa Ghaderi-Zefrehei3 and Mohsen Mardi2 |
| 1Department of Agronomy and Plant Breeding, Agricultural Science and Natural Resources University of Sari, Sari, Iran |
| 2Department of Genomics, Agricultural Biotechnology Research Institute of Iran (ABRII), Karaj, Iran |
| 3Department of Animal Science, University of Yasouj, Yasouj, Iran |
| *Corresponding authors: |
Arash Fazeli
Department of Agronomy and Plant Breeding
Agricultural Science and Natural Resources University of Sari
Sari, Iran
E-mail: arashfazeli57@gmail.com |
|
| |
| Received June 08, 2012; Published July 23, 2012 |
| |
| Citation: Fazeli A, Jelodar NB, Ehya F, Farsad LK, Ghaderi-Zefrehei M, et al. (2012) Development of DNA Microarray Technique to Evaluation of Transcriptional Responses of Wheat to Mycosphaerella graminicolla (septoria tritici). 1: 152. doi:10.4172/scientificreports.152 |
| |
| Copyright: © 2012 Fazeli A, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. |
| |
| Abstract |
| |
| We developed DNA microarray technique to study transcriptional responses of wheat to Mycosphaerella graminicola (septoria tritici). A set of 27 genes and fragments that involve in resistance to foliar disease in wheat and other cereals were amplified by PCR and printed in onto glass slides in six replicates. We carried out experiment in complete block design with three replications for two cultivars and inoculated plants with Mycospharella graminicolla fungi over four time points(6h, 6 day, 12 day and 18days). After inoculation, we collected samples from two cultivars. Total RNA was extracted from treatment and control samples and cDNA labeling were prepared by reverse transcription using dUTP and dUCP of all samples. Results indicated that four genes showed differential expression pattern between treatment vs control samples in Frontana cultivar. Annotations search indicated that these genes are involving in producing pathogen resistance proteins in plant. Hierarchical clustering and PCA analysis showed that these genes were separately clustered in seven groups and also, 32.94% of total variation described by two principle components in Frontana. |
| |
| Keywords |
| |
| Triticum aestivum; Mycosphaerella graminicola; DNA microarray; Resistance and susceptible wheat |
| |
| Introduction |
| |
| Bread wheat (Triticum aestivum) is one of the most important food crops in the world. In 2008, approximately 683 million tons wheat grain was produced from approximately 222 million hectares of land [1]. Wheat provides 21% of the food calories and 20% of the protein to more than 4.5 billion people in the 94 developing countries [2]. Septoria tritici blotch (STB), caused by the fungus Mycosphaerella graminicola (septoria tritici) is one of the most important foliar diseases of wheat in the worldwide that causing yield losses between 30 and 40 % [3]. Resistance cultivars and fungicides can be used to manage of STB. But, recently resistance to strobilurins fungicide is broken [4-9] and resistance level to azole fungicide increasing as well as [4-6,10]. So far, 18 Stb genes resistance have been identified that most of these genes have very low effect on resistance, therefore, breeding programs are only be used [11-14]. Microarrays are powerful tool for investigation of functions gene [15] that at present has become routine in many research laboratories. Various methods such as serial analysis of gene expression (SAGE) [16], differential display [17], Oligonucleotide arrays [18] and cDNA microarray [19] have been developed to show profile of gene expression over time, treatments and etc., in mRNA expression levels in large -scale assessments have been provided. The most common use of these techniques is to identify different patterns of gene expression or to compare mRNA expression levels between cells specific to different stimuli or different cellular phenotypes or stages of development. There are two types of microarray experiments include single and multiple-slide. In single- slides, transcript abundance of two mRNA samples are hybridized to the same slide but in multiple- slides, transcript abundance of two or more mRNA samples are hybridized to different slides [20]. DNA fragments, a set of genes that are amplified by PCR anmechanically spotted at high density on microscope slides using a robotic system, is relatively a simple x, y, z to a microarray containing thousands of pieces that are used [21]. Microarray can be created by inserting of DNA amplicon or cDNA clones on glass slide. Generally, amplicons length of PCR is in order of several hundred to thousand base pairs that have been used as an probe for each gene. This arrayer can be produced by researcher or company [18]. The microarrays are co-hybridization method using two or more fluorescently labeled probes that are made from RNA messenger from interest tissue [21]. The process of hybridization allows determination of relative expression level based on the ratio of each probe that hybridizes to an individual array. Hybridization measured using a confocal laser scanner to measure fluorescence intensity, allowing the simultaneous measurement of the relative level of expression of all genes present in the array. So far, microarray analysis for transcriptome changes in wheat again Mycosphaerella graminicolla has not been done but there are some publications on transcriptome response to biotic and abiotic stress in wheat by microarray technique such as Debbei et al. [22], Golkari et al. [23], Wang et al. [24], Qin et al. [25], Coram et al. [26], Xin et al. [27], Zarate et al. [28], Cao et al. [29] and also some scientist published article using cDNA-AFLP method to identify some resistance genes against septoria tritici in wheat such as Adhikari et al. [30], Sedaghatfar et al. [31]. The microarray study can be generally divided into three stages: (i) array fabrication; (ii) probe preparation and hybridization; and (iii) data collection, normalization and analysis (3). According to previous study of our colleagues identify 47 transcript-derived fragments (TDF) by cDNA-AFLP method between this two cultivars [31], Therefore, the main aim of this study was to identification of genes that show significant differential expression between two cultivars against Mycosphaerella graminicolla fungi based on resistance genes that have been used in our study. Also, understanding molecular functions and biological pathways of genes that have differential expression shaped the minor aim of this study. |
| |
| Materials and Methods |
| |
| Plant materials |
| |
| Two wheat cultivars, susceptible (Falat or Seri82) and resistant (Frontana) were used in this study. The susceptible cultivar used this study was Falat (Seri82) that is spring cultivar and produced in the CIMMYTE center. Frontana, a Brazilian Spring cultivar that is resistance to Fusarium head blight [32,33] has high resistance to the STB. All experiments were performed in a greenhouse at the Department of Genomics, Agricultural Biotechnology Research Institute, Iran (ABRII). |
| |
| Fungal inoculum and inoculation |
| |
| Firstly, in order to determinate aggressive of four isolates (MJA, SAL852-2, 19 Kermanshah and BL1S1) on two cultivars, we designed experiment for each isolate on two cultivars in complete block design with three replications. Inoculums were prepared from four isolates of M. graminicola that was collected from the Iran. The pure culture of the four isolates was revived by the culture the pieces isolate onto the Petri dish containing Potato Dextrose Agar (39g) of PDA dissolved in 1 liter water and autoclaves 121°C for 15 min then medium layout on the Petri dish and each isolate slowly layout on the medium by loop and keeping in incubator 18°C for three days. After that, Isolates that have been grown lodge in refrigerator 4°C to grow complete. By distillated water spores collected from the Petri dish and inoculums suspension adjusted to 1×107 spores/ml [34] by hemacytometer prior to inoculation. One drop of Tween 20 (polyoxyethy- lene-sorbitan monolaurate; Sigma- Aldrich, St. Louis, USA) was added per 100ml of spore suspension. Spores (~20ml/pot) were sprayed with a hand-operated sprayer onto each plant approximately 14 days after sowing. Inoculated plants were enclosed with polyethylene sheeting lined and misted with tap water to maintain near 100% relative humidity. 58 hours the polyethylene sheeting were removed and inoculated plants were left uncovered on the green- house benches. Greenhouse temperatures ranged from 20 to 24°C during the day (mean 22°C) and from 18 to 22°C at night (mean 20°C). The pots were watered daily or as needed and plants were scored for disease after symptoms developed (Figure 1). |
| |
|
|
Figure 1: Symptoms of Septoria tritici on leaves of two cultivars 24 days after infection (left) and leaf area infection in Falat (treatment vs control) 24 days after infection (right). |
|
| |
| Sampling procedure |
| |
| Evaluation aggressive of isolates in the greenhouse: 21 days after inoculations the result indicated that the SAL852-2 isolate has a good effect on infection of susceptible cultivar but there was not any symptom on resistance cultivar. Therefore, we designed a basic experiment in complete block design with SAL852-2 isolate and four times (6 hour, 6days, 12 days, 18days) [30]. After inoculation, we collected samples and immediately fixed on liquid nitrogen for further analysis. Each treatment consisted of mock inoculation with water (control) and inoculation with M. graminicola infected. For the timecourse infection assays, each treatment was sampled at 4 time points following Inoculation (6 h, 6D, 12D, 18DAI), generating 48 samples in total (two cultivars, 4 time points and three replications). Sampled plants were severed at the base of the stem with scissors and frozen immediately in liquid nitrogen. Three plants for each sample were harvested as biological replications, placed in a labeled plastic bag and stored at -80°C. RNA extractions were performed using RNA easy mini kit plant (QiaGene, Hilden, Germany) for all samples and quantitative and quality of RNA identified by spectrophotometer (NanoDrop 1000-Spectrophotometer, Thermo Scientific, USA) and agarose gel. The DNA concentration was measured by UV spectrophotometer for normalization in PCR reactions. To reduce the error in gene specific dye bias, we used Dye-swaps in one replication in our experiment [35]. |
| |
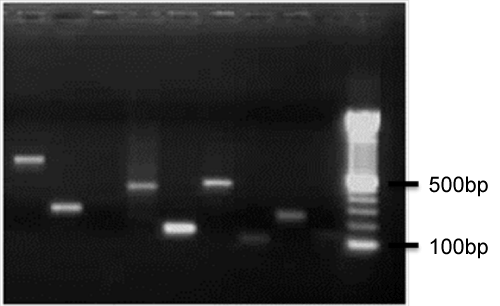
| Probe designing: In order to prepare of probe firstly we designed a primer by Oligo software v.5 for all sequences that we wanted to use in our experiment. these criteria were used in designing of primer: 18-22 bp long, melting temperature 50-65°C with 1-1.5°C differences between forward and reverse primers, 45-60% GC content, amplicon length 150-750bp. Secondly, sequences in database were Blasted to ensure that the primer amplification of areas that we are looking. After designing of primer, first, we tested primers on the DNA genomic of wheat to ensure that the length of the primers with the desired piece is identical and then PCR product of each primer were taken on 1% agarose gel. DNA genomics was extracted according Plant DNA Extraction Protocol for DArT (Diversity Array Technology, Australia). After this step we performed purification of PCR product using High Purification Kit (Roche, Mannheim, Germany), precipitated with ethanol and resolved to concentration of 0.1mg/ml and then distributed in 384 plate. Purified PCR product fixed on the amine arraying slide by Qarray robotic with 4 pines (Qarray system,genetix, UK) that pin touching slides two times per spot and printing six spot per gene. This PCR product printed on amine arraying slides that coated with amine reactive groups. Our microarrays have an 8×4 field layout with 32 spots per filed layout (192 spots on each slide) that each spot had a diameter of approximately 200 μm (Figure 2). |
| |
|
|
Figure 2: Results of PCR amplification of some genes on 1.5% agarose gel after purification. |
|
| |
| Array designing: Microarrays are prepared by printing PCR amplicons suspended on Amine arraying slide (Genetix) using high speed robotic system. This process first was presented by Patrick Brown and collaborators [19] at Stanford University and they provided scheme so that others can replicate arraying robot. However, there are many of companies that they are selling robotic systems for microarraying. We used Qarray robot system built by Genetix company. The microarray was designed on Amine arraying slide by Qarray system following the manufacture protocol. First we created the GAL file that shows position of PCR product on 384 plates. GAL file entered to robotic system. Concentration of PCR products (150-750 bp) of genes were 0.1-1 μg/μl and they are fixed on slides as a probe by robotic according to manufacture protocol. After that, slides were twice washed in 0.1% SDS and treated with blocking solution (0.75g succinic anhydride dissolved in 125 ml 1-methyle-2-pyrrolidinone then 125 ml 0.2M boric acid pH = 8 was added) for 20 minutes. Then slides were three times washed with dH2O at room temperature. In order to denature of double strand DNA, slides were incubated in dH2O at 95-100°C for 2 minutes then slide dried by placing them in slide dryer and centrifuged for 1 minute at 1000 rpm. Slides stored at room temperature for two weeks. |
| |
| Preparation of cDNA labeling: According to Cyscribe First-Starnd cDNA labeling kit (GE Healthcare, UK), the labeling cDNA were made using primer anchored oligo dT primer in the presence of both dUTP or dCTP nucleotide mix with dUTP or dCTP CyDye-labeled nucleotide. Each 11 μl reaction contained 2.5 μg of total RNA, 1μg of Anchored oligo dT and water incubated at 70°C for 5 minutes then it allowed the reaction to cool down on the ice and added labeling components in the following order; 5x CySribe buffer 4 μl, 0.1 M DDT 2 μl, dUTP or dCTP nucleotide mix 1 μl and CyScribe reverse transcriptase 1 μl that final reaction volume after addition of all components were 20 μl that mix reaction by vortex and centrifuge at 1000 rpm for 30s and incubated the reaction in 42°C for 1.5 hours and immediately purified the cDNA labeling CyScribe GFX purification kit (GE Healthcare UK) according to the protocol of manufacture. At the end, the labeled cDNA concentration was determined using microarray option in Nanodroop 1000 (NanoDrop 1000-Spectrophotometer, Thermo Scientific, USA) and stored cDNA labeling at -20°C in the amber tubes. |
| |
| Microarray hybridization and scanning: Equal volumes of cDNA labeling and genHYB (microarray Hybridization Buffer, Genetix) were mixed that 2.5-3 μl of final probe used per cm2 on slides (approximately 10μl for each slide) then final probe warmed up at 45°C for 2 minutes and quickly pipetted the appropriate volume of final probe on the arrayed area and gently lay on slides . Then, slides incubated in Genetix hybridization chamber at 42°C for 22 hours. After that, slides washed with composition of 1X SSC/0.2% SDS at 42-45°C for 5 minutes, 0.2X SSC/0.2% SDS at 42-45°C for 5 minutes and in the final 30s in 0.1XSSC at 42-45°C. After the final step of slide washing, slides dried by placing them in Genetix slide dryer and finally centrifuged at 1000 rpm for 1 minute. Slides were kept at the dark box to be away from dust until scanning. Slides were scanned at 5 μm resolution by ScannArray Express (Perkin Elmer, USA). The same laser power (90%) was used for all slides. |
| |
| Statistical analysis: Data analysis carried out using Bioconductor (www.bioconductor.org) in the R (2.13.1) program. First, we calculated the average median of six replications of each spot on slid as a median intensity for each gene. The spot intensity of each gene was calculated as the median foreground intensity of the spot minus the median background intensity around the spot. Pearson correlation coefficients were calculated for log2 fold differences within slide and between the same spot on different slides with raw and normalized intensity for both experiments. The fold changes for normalized data were estimated by fitting linear model using the limma package (release 1.8.1). To show which RNA samples were applied to which array, after that, we constructed design matrix for each experiment (control vs treatment) and made contrast four times in each experiments. For identifying differential transcript abundance, we used limma and empirical Bayes method to moderate standard errors of log fold-changes. Moderated t-statistic used to identify significance of the differential expression. Moderated t-statistic created p-values although degrees of freedom have been increased that this indicated more validation with the smoothed standard errors. In order to account for multiplicity (multiple comparisons), p-values adjusted and in this way, we used Benjamini and Hochbergs [36] method to calculated the false discovery rate (FDR). FDR adjusted p-values ≤ 0.1 (cut off value) was applied to give <1% false positives. Also, we performed PCA (principle component analysis) and hierarchical clustering to show relationship between genes using R (2.13.1). |
| |
| Results |
| |
| Disease incidence |
| |
| Our Primary results indicated that among four isolates that have been tested on two cultivars, SA852-2 isolate had more Pathogenesis on the susceptible cultivar (Seri 82), in contrast, there were no symptoms on resistance cultivar (Frontana). Seri 82 was highly susceptible to this isolate and first lesions at principle exam were visible 15 DPI after contamination on Seri82. Disease severity as percentage of leaf area with infection increased rapidly afterwards and at 24 DAI about 80% of leaf area infected in Seri82. At this time, all asexual spores produced a high density of lesions that shown by pycnidia. In contrast, there was not observed significant necrosis or discoloration in Frontana even at 27 DPI. |
| |
| Preparation of cDNA microarray and RNA samples |
| |
| |
| For initiation and development of microarrays, we first identified 27 genes from public database (Table 1). The selected genes encode enzymes involved in many different aspects of resistance to disease such as pathogenesis related protein (PRP), mitogen activated protein kinase (MAPK) and lipid transfer protein precursor in cereals. Portion of these genes were amplified from genomics DNA by PCR, purified and printed in six replicates on aldehied amine coated microscope slides. To check whether expression of these genes altered during different treatments, we carried out our experiment in complete block design with three replications with SA852-2 isolate and four time points after infected seedling leaves harvested and used for microarray analysis using three biological replications and total 48 arrays were hybridized. Total RNA extraction from the leaves and qualities of RNA has good effect on cDNA synthesis and all downstream steps in the analysis of gene expression. High-quality RNA from powdered leaves was extracted according manufacture protocol (Qiagen, Hilden,Germany). After that, their quality and quantity got checked by Mops gel and nanodroop 1000 (NanoDrop 1000-Spectrophotometer, Thermo Scientific, USA) respectively. cDNA synthesis from total RNA using anchored oligo-dT kit (GE Healthcare, UK) and after that quality and quantity of cDNA was checked by (NanoDrop 1000-Spectrophotometer, Thermo Scientific, USA) and graphs from the wavelength absorption indicated that quality and quantity of graphs for each dye is as it was expected (data not shown). RNA isolated from the treatments samples was labeled with the florescent dye cy5 that show wavelength emission spectra at 650 nm, in contrast, RNA isolated from untreatment (control) samples was labeled with the florescent dye cy3 that wavelength emission spectra is 550 nm . We used substitution of dyes in one replicate at each experiment at two conditions (treatment and untreatment) to control or reduce error by dye bias [35]. The cy3 and cy5 labeling cDNA were mixed and hybridized to microarray slides. After hybridization and washing, we normalized total cy3 and cy5 signals using the scanner normalization factor. In general, microarray data analysis begins with data normalization to reduce the experimental variations due to changes in the spot location on different arrays and maintain biological variations [37]. There are several statistical methods to normalize microarray data [38]. And also, there are several statistical methods to calculate the relative gene expression from normalized microarray data including t tests, non-parametric tests and Bayesian models [39]. Common goal of different microarray data normalization methods, is to identify or remove systematic source variation (e.g. Efficiency of different labeling, scanning properties of dyes and print tip). |
| |
|
|
Table 1: Primers and genes that printed on microarrays used in this study. |
|
| |
| Analysis: normalized weighted average log data set |
| |
| The spot intensity for each channel (cy3 and cy5) has a single value associated with channel intensity minus background intensity was normalized. Our average of total signal intensity for each gene was as an average of the total intensity (the sum of the measured intensity of theCy3 and Cy5 channels) of the six spots representing the gene. Logarithm of the normalized values (LNV) of each channel for each spot was taken and propagating errors through the log transformation: |
| |
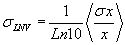 |
| |
| For all slides, using the method maximum likelihood, the LNV for each gene and each channel as a weighted average for each gene was calculated of mean each gene. |
| |
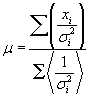 |
| |
| Which data point of xi in the sum is weighted inversely by its variance σ2. SD-weighted average is calculated using the following formula: |
| |
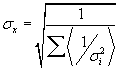 |
| |
| Identification of Differential Expressed Genes |
| |
| Statistical analysis showed that our microarray preparation and scanning method was good. Based on quality confidence calculation of spots intensity, we deleted outliers from the genes to distinguish those genes expression which were significantly from the mean. We used easy quant protocol to calculate spot intensity (Adaptive circle) based on quantitation method of slides by scanner. The data set from each slides were combined to produce a single data set for each cultivar. Analysis of this data using Bioconductor (www.bioconductor.org) in the R (2.13.1) software for two cultivars and related controls in four times indicated that some genes, based on mean intensity of each spot (Table 1), had different expression patterns between two conditions (control vs treatment) in Frontana Results at Table 2 show that five genes are differentially expressed on four time points in Frontana (resistance) in comparing with controls that most of genes expressed early times after infection in Frontana. The PRP3 and PR1 genes at early times (6h and 12 h) showed different expression pattern in Frontana compared with control. Also, three genes i.e. PRP1, Ppi and Brox in 5% cut off vlaue showed significant differential expression in 12 Days after infection in Frontana (resistance)in comparing with control at this time. Other resistances genes maybe involve in resistance to fungi but they did not show significant expression between treatment and untreatment samples at different times in our experiment. In contrast, in susceptible cultivar none of the studied genes showed significant expression between treatment and untreatment in four times. Our observation in greenhouse indicated that SA852-2 isolate had good effect on infection of susceptible cultivar but cannot affect Frontana. Therefore, the disease severity 24 Days after infection in Seri 82 was 80%. This shows that differences between resistance and susceptible cultivar may be due to the expression of several genes. In our study, some of them had different expression pattern. Based upon our results, none of the resistance genes are expressed in the susceptible cultivar. Resistance of Frontana cultivar against Mycosphaerella graminicolla fungi, reflected in gene expression, changes by these fungi. |
| |
|
|
Table 2: Statistical parameter for genes that showed differential expression in four time points. |
|
| |
| Hierarchical Clustering and PCA Analysis |
| |
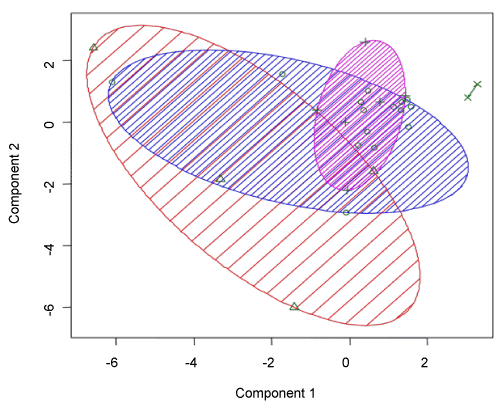
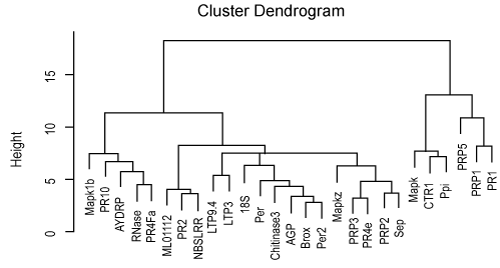
| To evaluate the relationship between genes in two cultivars, we analyzed linkage hierarchical clustering using 27 genes for Frontana that showed differential expression in genes at two conditions using pvclust package in R softwar. This package provides p-value for hierarchical clustering based on multi-scale bootstrap re-sampling (Figure 3). Analysis of hierarchical clustering in Frontana cultivar was done on intensity expression of genes at two conditions indicated that genes divided in four groups. Interesting genes that showed significant expression in microarray experiment in Frontana cultivar are being clustered in the same group. However, the Ppi gene is being clustered in the other subgroup. The PR1, PRP1 and PRP5 genes were clearly separated from the other genes in Frontana. Also, PR1 and PRP1 that showed significant differential expression in 1% cut off value, located in the same subgroup but PRP5 which showed differential expression pattern at the 5% of cut off is being located in other subgroup. Other genes which were significant at the 5% cut off value, were grouped in different subgroups. Principle component analysis (PCA) also indicated that 32.93 of total variation explained by two first components (Figure 4). |
| |
|
|
Figure 3: Principle component analysis (PCA) for Frontana cultivar. |
|
| |
|
|
Figure 4: Average linkage hierarchical clustering analysis of the log2 transformed fold changed ratio of the 27 Genes in Frontana. |
|
| |
| Major functional categories of genes in wheat |
| |
| Annotation of genes that we used in our study summarized in Table 1. However, genes that identified by Adhikari et al. [30] involved in many different aspecst of resistance to M. graminicolla fungi. Our further analysis was focused on function of these genes in database. Most of them involved in resistance to pathogen such as pathogenesis related protein, mitogen-activated protein kinase and disease resistance protein. PR1, PRP1 and PRP5 genes that showed significant different expression in microarray experiment belonged to pathogenesis related protein. |
| |
| Pathogenesis related protein (PRP) |
| |
| Pathogenesis related (PR) protein associated with disease resistance in several plant-pathogen interactions have been investigated [40,41]. Function of some of them PR –proteins are unknown [41,42], while others are involved in antifungal activity in vivo PR2 (1,3 β-glucanase) and PR3 (chitinase) degraded cell walls and may be directly inhibit growth pathogen [43,44]. PR2 and PR3 released from the hydrolysis of fungal cell wall and act as elicitor in defense reaction [45-47] and so serve as PAMPs/MAMPs [48,49]. Recognition of PAMPs and MAMPs activated defense reaction in plant. These responses include accumulation of Reactive Oxygen Species (ROS) and PR-proteins that reinforcement of the cell wall by oxidative cross-linking of these components and deposition of callose and lignin [48,50]. Synthesis of callose and β-1, 3 –glucanase occurs de novo as a response to pathogen attack [51-53]. |
| |
| Using differentially display polymerase chain reaction (DD-PCR), Ray et al. [54] identified pathogenesis-related (PR) protein (PR2) which showed the early and strong resistance-related response. Similarity of this gene with other 1,3 β-glucanase was 96% and highest transcript level of this gene observed 6 and 12 h after inoculation with the pathogen. Adhikari et al. [30] showed that induction of PR1 was 10- 60 folds at early stage during incompatible interaction between host and pathogen. Ray et al. [54] based on expression profiles during at four first days identified three pathogenesis related (PR) proteins PR1, PR2 and PR3 which were much more induced at 3-12h in resistance wheat leaves (Tadiana) after they were inoculated. Precisely how these genes participate in immune response is not clear, but different roles have been proposed [55]. Production of toxic intermediate PR proteins inhibits the germination of spores and growth of pathogen [56]. Ray et al. [54] concluded that the outcome of the host-pathogen interaction will be determined over 24 h after infection. Shetty et al. [57], found that resistance again M. graminicola is dependent on the early accumulation of β-1,3-glucanase which may directly inhibit the pathogen and protect host against fungal enzymes and toxins. Peptidyl-prolyl isomerases (Ppi) proteins that are expressed in prokaryotic and eukaryotic cells and so far, three classes of Ppi have been identified [58]. In this study, we used the wheat gene that based on BLAST had highest similarity with to the FK506-binding proteins which are also known as immunophilins. These proteins with chaperons like heat-shock protein (Hsp) 70 and Hsp 90 created a complex that involved in steroid pathway signaling [59]. Ray and colleagues [54] showed that most defense-related genes in two cultivars’ resistance (Tadinia and W7984) after infection associated with signaling, energy metabolism and protein synthesis. More induction of Ppi gene in resistance cultivars than susceptible cultivars indicated that both energy production and signaling pathway hormones are more active in resistance cultivars than susceptible. Adhikari et al. [30] showed that the activation of these genes in resistance cultivars when the fungal biomass increased exponentially in susceptible cultivars. Therefore, it can be concluded that these genes are necessary for defensive responsive against Mycosphaerella graminicolla at late time. Serine CarboxyPeptidase (SCP) in higher plant interacellular enzymes that have N-terminal signal peptides that localized in vacuoles and they functioned in intracellular protein turnover [60,61]. In mammals, Scp known as “protein protection” that can protect other enzyme from degradation in lysosome [61]. Our results indicated that Scp gene have significant expression in comparing with control in Fronatna at 6 DAI. Some publication indicated that Scp have different role in plant: disease resistance [30,62] secondary metabolism [63] degradation of the protein synthesis antibiotic [64]. Adhikari et al. [30] using cDNAAFLP method showed that Scp gene in Tadinia (Resistance cultivar) had different pattern expression at 3h, 1 and 6 DAI, while in the other cultivar it showed a little change in expression profile. Mugford et al. [62] identified Saponin-deficient 7 locus (Sad7) as being required for the synthesis of antimicrobial triterpene glycosides (avenacins) that involved in the broad-spectrum disease resistance in diploid oat (Avena strigosa). This locus encodes a functional Serine carboxypeptidaselike (SCPL) protein that is able to catalyze the synthesis of both N-methyl anthraniloyl- and benzoyl-derivatized forms of avenacin. The economic damage by M. graminicolla in the world makes it necessary to develop effective method to control of disease; current methods include use of fungicide and resistance cultivar [4-6] that have partial resistance to septoria tritici. Here, we described technique that should further experiment with a lot of genes or entire genome sequence of wheat (Triticum aestivum) that is not known. Microarray can provide statistically significant data for understanding the interplay of known genes in controlling biotic or abiotic stresses in wheat or other crops. Table 2 showed the genes that have significant expression in treatment vs control in our study. Among five genes that showed different expression, three themare associated with pathogenesis related protein (PRP). Antoniw et al. [65] coined term Pathogenesis related protein (PRs) that defined as “proteins encoded by the host plant but induced only in pathological or related situations” that implying of non-pathogenic origin. PR1 with Thaumatin- like protein have antifungal activity in combination with a wide range of virulence and avirulance fungi. Increasing of information about function of PR proteins such as disease resistance, developmental and adaption to stressful environment, encourage researcher to apply PR genes in gene- engineering technology to improvement plants and produce plants that have high adaption to stress condition. Empirical evidence is needed to prove the utility of the PRs genes to the development of disease resistance in transgenic plant. The practical aspects of PRs genes resulted in ergonomically important crops resistance to various disease. Results from transgenic plant such as: over expression of PR2 and PR3 in potato transgenic enhance resistance to Phytophthora infestans [66], Brassica napus [67], rice ryegrass [68], rice [69] and tobacco [70]. |
| |
| Acknowledgment |
| |
| We gratefully acknowledge the Genomics Department of Agricultural Biotechnology Research Institute of Iran (ABRII) due to financial supporting and Dr. Ezatollah Sedaghatfar for helping during this project. |
| |
| |
| References |
| |
- https://faostat.fao.org/site/567/DesktopDefault.aspx?PageID=567, 02 September 2010
- Braun HJ, Atlin G, Payne T (2010) Multi-location testing as a tool to identify plant response to global climate change. Climate change and crop protection.
- Eyal Z, Scharen AL, Prescott JM, van Ginkel M (1987) The Septoria diseases of wheat: Concepts and methods of disease management. CIMMYT: 46.
- Cools HJ, Fraaije BA (2008) Are azole fungicides losing ground against Septoria wheat disease? Resistance mechanisms in Mycosphaerella graminicola. Pest Manag Sci 64: 681-684.
- Fraaije BA, Cools HJ, Fountaine J, Lovell DJ, Motteram J, et al. (2005) Role of Ascospores in Further Spread of QoI-Resistant Cytochrome b Alleles (G143A) in Field Populations of Mycosphaerella graminicola. Phytopathology 95: 933-941.
- Fraaije BA, Cools HJ, Kim SH, Motteram J, Clark WS, et al. (2007) A novel substitution I381V in the sterol 14alpha-demethylase (CYP51) of Mycosphaerella graminicola is differentially selected by azole fungicides. Mol Plant Pathol 8: 245-254.
- McCartney C, Mercer PC, Cooke LR, Fraaije BA (2007) Effects of a strobilurin-based spray programme on disease control, green leaf area, yield and development of fungicide-resistance in Mycosphaerella graminicola in Northern Ireland. Crop Protect 26: 1272-1280
- Stammler G, Carstensen M, Koch A, Semar M, Strobel D, et al. (2008) Frequency of different CYP51-haplotypes of Mycosphaerella graminicola and their impact on epoxiconazolesensitivity and -field efficacy. Crop Protect 27: 1448-1456
- Torriani SF, Brunner PC, McDonald BA, Sierotzki H (2009) QoI resistance emerged independently at least 4 times in European populations of Mycosphaerella graminicola. Pest Manag Sci 65: 155-162.
- Mavroeidi VI, Shaw MW (2005) Sensitivity distributions and crossresistance patterns of Mycosphaerella graminicola to fluquinconazole, prochloraz and azoxystrobin over a period of 9 years. Crop Protect 24: 259-266
- Ghaffary SM, Robert O, Laurent V, Lonnet P, Margalé E, et al. (2011) Genetic analysis of resistance to septoria tritici blotch in the French winter wheat cultivars Balance and Apache. Theor Appl Genet 123: 741-754.
- Arraiano LS, Chartrain L, Bossolini E, Slatter HN, Keller B, et al. (2007) A gene in European wheat cultivars for resistance to an African isolate of Mycosphaerella graminicola. Plant Pathol 56: 73-78.
- Chartrain L, Sourdille P, Bernard M, Brown JKM (2009) Identification and location of Stb9, a gene for resistance to septoria tritici blotch in wheat cultivars Courtot and Tonic. Plant Pathol 58: 547-555.
- Goodwin SB (2007) Back to basics and beyond: increasing the level of resistance to Septoria tritici blotch in wheat. Australas Plant Pathol 36: 532-538.
- Nguyen DH, D'haeseleer P (2006) Deciphering principles of transcription regulation in eukaryotic genomes. Mol Syst Biol 2: 2006.
- Velculescu VE, Zhang L, Vogelstein B, Kinzler KW (1995) Serial analysis of gene expression. Science 270: 484-487.
- Liang P, Pardee AB (1992) Differential display of eukaryotic messenger RNA by means of the polymerase chain reaction. Science 257: 967-971.
- Lockhart DJ, Dong H, Byrne MC, Follettie MT, Gallo MV, et al. (1996) Expression monitoring by hybridization to high-density oligonucleotide arrays. Nat Biotechnol 14: 1675-1680.
- Schena M, Shalon D, Davis RW, Brown PO (1995) Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 270: 467-470.
- Dudoit S, Yang YH, Callow MJ, Speed T (2000) Statistical methods for identifying differentially expressed genes in replicated cDNA microarray experiments.
- Shalon D, Smith SJ, Brown PO (1996) A DNA microarray system for analyzing complex DNA samples using two-color fluorescent probe hybridization. Genome Res 6: 639-645.
- Laudencia-Chingcuanco DL, Stamova BS, You FM, Lazo GR, Beckles DM, et al. (2007) Transcriptional profiling of wheat caryopsis development using cDNA microarrays. Plant Mol Biol 63: 651-668.
- Golkari S, Gilbert J, Prashar S, Procunier JD (2007) Microarray analysis of Fusarium graminearum-induced wheat genes: identification of organ-specific and differentially expressed genes. Plant Biotechnol J 5: 38-49.
- Wang X, Tang C, Zhang G, Li Y, Wang C, et al. (2009) cDNA-AFLP analysis reveals differential gene expression in compatible teraction of wheat challenged with Puccinia striiformis f. sp. Tritici. BMC Genomics 10: 289.
- Qin D, Wu H, Peng H, Yao Y, Ni Z, et al. (2008) Heat stress-responsive transcriptome analysis in heat susceptible and tolerant wheat (Triticum aestivum L.) by using Wheat Genome Array. BMC Genomics 9: 432.
- Coram TE, Settles ML, Chen X (2009) Large-scale analysis of antisense transcription in wheat using the Affymetrix GeneChip Wheat Genome Array. BMC Genomics 10: 253.
- Xin M, Wang Y, Yao Y, Song N, Hu Z, et al. (2011) Identification and characterization of wheat long non-protein coding RNAs responsive to powdery mildew infection and heat stress by using microarray analysis and SBS sequencing. BMC plant biology 11: 61.
- Zárate X, Henderson DC, Phillips KC, Lake AD, Galbraith DW (2010) Development of high-yield autofluorescent protein microarrays using hybrid cell-free expression with combined Escherichia coli S30 and wheat germ extracts. Proteome Sci 8: 32.
- Cao A, Xing L, Wang X, Yang X, Wang W, et al. (2011) Serine/threonine kinase gene Stpk-V, a key member of powdery mildew resistance gene Pm21, confers powdery mildew resistance in wheat. Proc Natl Acad Sci U S A 108: 7727-7732.
- Adhikari TB, Cavaletto JR, Dubcovsky J, Gieco JO, SchlatterAR, et al. (2004) Molecular mapping of the Stb4 gene for resistance to Septoria tritici blotch in wheat. Phytopathology 94: 1198-1206
- Sedaghatfar E, Zamanizadeh HR, Roohparvar R, Karimi Farsad L, Fazeli A, et al. (2012) Gene expression profiling of defense-related genes resistant to Septoria tritici blotch in wheat. African Journal of Biotechnology.
- Steiner B, Lemmens M, Griesser M, Scholz U, Schondelmaier J, et al. (2004) Molecular mapping of resistance to Fusarium head blight in the spring wheat cultivar Frontana. Theor Appl Genet 109: 215-224.
- Mardi M, Pazouki L, Delavar H, Kazemi MB, Ghareyazie B, et al. (2006) QTL analysis of resistance to Fusarium head blight in wheat using a 'Frontana'-derived population. Plant Breed 125: 313-317.
- Kema GH, van Silfhout CH (1997) Genetic Variation for Virulence and Resistance in the Wheat-Mycosphaerella graminicola Pathosystem III. Comparative Seedling and Adult Plant Experiments. Phytopathology 87: 266-272.
- Martin-Magniette ML, Aubert J, Cabannes E, Daudin JJ (2005) Evaluation of the gene-specific dye bias in cDNA microarray experiments. Bioinformatics 21: 1995-2000.
- Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Statist Soc B 57: 289-300.
- Hegde P, Qi R, Abernathy K, Gay C, Dharap S, et al. (2000) A concise guide to cDNA microarray analysis. Biotechniques 29: 548-550, 552-4, 556 passim.
- Tseng GC, Oh MK, Rohlin L, Liao JC, Wong WH (2001) Issues in cDNA microarray analysis: quality filtering, channel normalization, models of variations and assessment of gene effects. Nucleic Acids Res 29: 2549-2557.
- Efron B, Tibshirani R (2002) Empirical bayes methods and false discovery rates for microarrays. Genet Epidemiol 23: 70-86.
- Linthorst HJM (1991) Pathogenesis-related proteins of plants. Critical Reviews in Plant Sciences 10: 123-150.
- Van Loon LC, Van Strien EA (1999) The families of pathogenesis related proteins, their activities, and comparative analysis of PR-1 type proteins. Physiological and Molecular Plant Pathology 55: 85-97.
- Kitajima S, Sato F (1999) Plant pathogenesis-related proteins: molecular mechanisms of gene expression and protein function. J Biochem 125: 1-8.
- Kim YJ, Hwang BK (1997) Isolation of a basic 34 kiloDalton ß-1,3-glucanase with inhibitory activity against Phytophthora capsici from pepper stems. Physiological and Molecular Plant Pathology 50: 103-115.
- Kini KR, Vasanthi NS, Shetty HS (2000) Induction of ß-1,3-glucanase in seedlings of pearl millet in response to infection by Sclerospora graminicola. European Journal of Plant Pathology 106: 267-274.
- Takeuchi Y, Yoshikawa M, Takeba G, Tanaka K, Shibata D, et al. (1990) Molecular Cloning and Ethylene Induction of mRNA Encoding a Phytoalexin Elicitor-Releasing Factor, beta-1,3-Endoglucanase, in Soybean. Plant Physiol 93: 673-682.
- Wu G, Shortt BJ, Lawrence EB, Leon J, Fitzsimmons KC, et al. (1997) Activation of Host Defense Mechanisms by Elevated Production of H2O2 in Transgenic Plants. Plant Physiol 115: 427-435.
- Jia Y, Martin GB (1999) Rapid transcript accumulation of pathogenesis-related genes during an incompatible interaction in bacterial speck disease-resistant tomato plants. Plant Mol Biol 40: 455-465.
- Nürnberger T, Brunner F, Kemmerling B, Piater L (2004) Innate immunity in plants and animals: striking similarities and obvious differences. Immunol Rev 198: 249-266.
- Altenbach D, Robatzek S (2007) Pattern recognition receptors: from the cell surface to intracellular dynamics. Mol Plant Microbe Interact 20: 1031-1039.
- Göhre V, Robatzek S (2008) Breaking the barriers: microbial effector molecules subvert plant immunity. Annu Rev Phytopathol 46: 189-215.
- Skou JP, Jørgensen JH, Lilholt U (1984) Comparitive studies on callose formation in powdery mildew compatible and incompatible barley. Phytopathologische Zeitschrift 109: 147-168.
- Enkerli K, Hahn MG, Mims CW (1997) Immunogold localization of callose and other plant cell wall components in soybean roots infected with the oomycete Phytophthora sojae. Canadian Journal of Botany 75: 1509-1517.
- Verma DP, Hong Z (2001) Plant callose synthase complexes. Plant Mol Biol 47: 693-701.
- Ray S, Anderson JM, Urmeev FI, Goodwin SB (2003) Rapid induction of a protein disulfide isomerase and defense-related genes in wheat in response to the hemibiotrophic fungal pathogen Mycosphaerella graminicola. Plant Mol Biol 53: 701-714.
- Ride JP (1983) Cell walls and other structural barriers in defense. Biochemical plant pathology 215-236.
- Sutherland MW (1991) The generation of oxygen radicals during host plant responses to infection. Physiol Mol Plant Pathol 39: 79-93.
- Shetty NP, Jensen JD, Knudsen A, Finnie C, Geshi N, et al. (2009) Effects of beta-1,3-glucan from Septoria tritici on structural defence responses in wheat. J Exp Bot 60: 4287-4300.
- Shaw PE (2002) Peptidyl-prolyl isomerases: a new twist to transcription. EMBO Rep 3: 521-526.
- Riggs DL, Roberts PJ, Chirillo SC, Cheung-Flynn J, Prapapanich V, et al. (2003) The Hsp90-binding peptidylprolyl isomerase FKBP52 potentiates glucocorticoid signaling in vivo. EMBO J 22: 1158-1167.
- Zuber H, Matile P (1968) Acid carboxypeptidases: their occurrence in plants, intracellular distribution and possible function. Z Naturforsch B 23: 663-665.
- Rawlings ND, Barrett AJ (1994) Families of serine peptidases. Methods Enzymol 244: 19-61.
- Mugford ST, Xi X, Bakht S, Hill L, Wegel E, et al. (2009) A Serine Carboxypeptidase-Like Acyltransferase Is Required for Synthesis of Antimicrobial Compounds and Disease Resistance in Oats. The plant cell 21: 2473-2484.
- Lehfeldt C, Shirley AM, Meyer K, Ruegger MO, Cusumano JC, et al. (2000) Cloning of the SNG1 gene of Arabidopsis reveals a role for a serine carboxypeptidase-like protein as an acyltransferase in secondary metabolism. Plant Cell 12: 1295-1306.
- Agarwal V, Tikhonov A, Metlitskaya A, Severinov K, Nair SK (2012) Structure and function of a serine carboxypeptidase adapted for degradation of the protein synthesis antibiotic microcin C7. Proc Natl Acad Sci U S A 109: 4425-4430.
- Antoniw JF, Ritter CE, Pierpoint WS, Van Loon LC (1980) Comparison of three Pathogenesis- related proteins from plants of two cultivars of tobacco infected with TMV. J Gen Virol 47: 79-87.
- Bachmann D, Rezzonico E, Retelska D, Chételat A, Schaerer S, et al. (1998) Improvement of potato resistance to Phytophthora infestans by overexpressing antifungal hydrolases. 5th International Workshop on pathogenesis-related proteins.
- Grison R, Grezes-Besset B, Schneider M, Lucante N, Olsen L, et al. (1996) Field tolerance to fungal pathogens of Brassica napus constitutively expressing a chimeric chitinase gene. Nat Biotechnol 14: 643-646.
- Takahashi W, Fujimori M, Miura Y, Komatsu T, Nishizawa Y, et al. (2005) Increased resistance to crown rust disease in transgenic Italian ryegrass (Lolium multiflorum Lam.) expressing the rice chitinase gene. Plant Cell Rep 23: 811-818.
- Datta K, Velazhahan R, Oliva N, Ona I, Mew T, et al. (1999) Over-expression of the cloned rice thaumatin-like protein (PR-5) gene in transgenic rice plants enhances environmental friendly resistance to Rhizoctonia solani causing sheath blight disease. Theor Appl Genetics 98: 1138-1145.
- Sarowar S, Kim YJ, Kim EN, Kim KD, Hwang BK, et al. (2005) Overexpression of a pepper basic pathogenesis-related protein 1 gene in tobacco plants enhances resistance to heavy metal and pathogen stresses. Plant Cell Rep 24: 216-224.
|
| |
| |