Study of in vitro Therapeutic Equivalence of the 5 mg Glibenclamide Multi-source Tablets Respecting the Reference Medicine Product
Received: 06-Dec-2017 / Accepted Date: 11-Dec-2017 / Published Date: 16-Dec-2017 DOI: 10.4172/2167-065X.1000180
Abstract
Introduction: Therapeutic equivalence of medications is carried out through in vitro and in vivo studies called bioequivalence studies.
Objective: To determine the in vitro therapeutic equivalence of the 5 mg glibenclamide multi-source tablets respecting the reference medicine.
Materials and methods: Both, the multi-source drug 5 mg glibenclamide and the reference 5 mg Glidiabet are made in Peru, and were acquired in a drugstore of Ica city (Peru). Reagents and an analytical grade standard were used. The Ultraviolet absorption method at 300 nm was used on each of the three dissolution media.
Results: Neither the multi-source drug T nor the reference R dissolved by 85% at pH 1.2 and at pH 4.5, during 15 or 30 minutes. However, at pH 6.8 dissolution occurs. These results correspond to Food and Drug Administration and United States Pharmacopoeia criteria. The similarity factor value was within the acceptance range (50-100) for the three tested pHs. Dissolution efficiency was 68.66% (pH 1.2), 56.59% (pH 4.5) and 95.98% (pH 6.8). The mean of in vitro dissolution time was 28.56 min (pH 1.2), 39.97 min (pH 4.5) and 4.54 min (pH 6.8).
Conclusion: According to the similarity factor (f2) and the efficiency of dissolution, it is concluded that the multisource drug 5 mg glibenclamide of the present study is therapeutically equivalent in vitro to the reference 5 mg Glidiabet.
Keywords: In vitro therapeutic equivalence; Dissolution profile; Multi-source drug T; Similarity factor; Dissolution efficiency
Introduction
The therapeutic equivalence studies allow the demonstration of the bioequivalence and interchange ability of the multi-source drugs, for this reason, relative bioavailability studies or in vitro bioequivalence studies must be done [1,2].
Studies of therapeutic equivalence in vitro are tests that consist of comparing the dissolution kinetics of the multi-source drug with the referent, the same that is carried out in three dissolution media at pH 1.2; pH 4.5 and pH 6.8; which allows establishing in vitro and in vivo correlations. This study is carried out based on the bio-exclusion criteria according to the Biopharmaceutical Classification System (BSC): [3,4] Class 1 drugs (high solubility and high permeability) must be dissolved in more than 85% in 30 minutes, it is considered highly soluble, when the drug in its highest dose is soluble in 250 ml or less of aqueous medium in a pH range of 1.2-7.5, according to the FDA, and of 1.2-6.8, according to the WHO. A drug is considered highly permeable (degree of absorption), if the amount absorbed is greater than 85%, according to the WHO, and 90% according to the FDA,4 being the solution, a factor limiting the rate of absorption; 5 Class 2 (of low solubility and high permeability) must be weak acid drugs, whose absorption is limited by the rate of dissolution,6 and must dissolve rapidly in more than 85% at a pH of 6.8 in 30 minutes or less, and Class 3 (high solubility and low permeability) must dissolve quickly in more than 85% in 15 minutes or less at pH 1,2, 4,5 and 6,8 simulate the middle of the gastrointestinal tract; In these drugs, the limiting step in the rate of absorption is permeability and not dissolution. While Class 4 (low solubility and low permeability), are not accepted from the realization of equivalence studies in vivo, since it is difficult to obtain in vitro and in vivo correlations [5-10].
The kinetics of dissolution depends on technological and formulation factors, which may be dependent on the solid to be dissolved: a) drug (physicochemical characteristics: solubility that depends on the chemical nature, chemical state of salt, acid, base or ester, polymorphism, impurities ); b) free surface: particle size and porosity; dependent on the pharmaceutical form (technology used for its manufacture, wetting capacity, penetration capacity in the dissolution medium, type and quantity of excipients in the formulation such as disintegrants (starch, sodium glycolate-starch), binders (polyvinyl pyrrolidone, derivatives of cellulose and corn starch), diluents (lactose, glucose and microcrystalline cellulose), lubricants (magnesium stearate, stearic acid, polyethylene glycol, sodium chloride and talc) and surfactants, packing and storage conditions. they depend on the dissolution medium as the composition of the medium (pH, viscosity, surface tension, presence of adsorbents and salts), intensity of agitation and temperature [11,12].
The comparison of the dissolution profiles is done using the independent model of the difference factor (f1), which measures the percentage of error between the two curves in the sampling time periods. The results are interpreted based on values: if the value is zero, it indicates that the two curves are equal, if the value is from 0 to 15 the two curves are similar, and if the value is greater than 15 the profiles are different [13]. The second method is the independent model of the similarity factor (f2), proposed by Moore and Flanner, which is the logarithmic transformation of the reciprocal of the square root of the sum of the squared errors and is a measure of the similarity between the two dissolution curves. It is calculated from the difference between the average values of the dissolved percentage between the multi-source medicine and the referent [5]. To ensure the equivalence of the two curves of the dissolution kinetics, the value must be 100, indicating that the curves of the drug test are identical to the referent, if the value is 50 to 100 the similar, 4, 5, 13 if the value is equal to 49,89 (rounded to 50) it is concluded that there is a difference of 10% in the curves [5].
The dissolution efficiency (EF%), is the area under the curve of dissolution at a time (t), expressed as a percentage of the rectangle described for the 100% dissolution in the same time, so, the resulting value must be greater than 50% for immediate-release medications. The degree of absorption of a drug “in vivo” is proportional to the concentration of the drug in the solution and at the same time the contact with the gastrointestinal mucosa where it is absorbed, so it is described as a function of these two variables. The mean dissolution time (MDT) is the residence time of the drug in the solid state in a solution [9,13,14].
Glibenclamide is an acidic drug, whose chemical name is 1-[4-[2-(5-chloro-2-methoxybenzamide) ethyl] benzenesulfonyl] -3-cyclohexylurea, and according to the BCS it is class 2 [15,16]. This is a second generation sulfonylurea, used in the treatment of Diabetes mellitus type 2. Its dissolution kinetics depends on the pKa 5.3 of the molecule, the solid dispersion, the surfactants, the media [17-20] that simulate being with food and polymorphisms. There are four forms of polymorphs, form I is simple triclinic, form II is orthorhombic, form III monoclinically simple, and IV is monoclinic Centered at the base, being the most used form I and III [21]. The pharmacokinetic parameters are: maximum time (tmax) of 4 hours, protein binding (UP) by 99%, half-life (t1/2) of 10 hours and an apparent volume distribution (Vd) of 0.3 L/kg. It is metabolized by 3-cis and 4-trans hydroxylation of the cyclohexyl ring [18].
In Peru there is little information on the studies of dissolution profiles or in vitro therapeutic equivalence, despite having a wide variety of multi-source (generic and similar) and innovative drugs. There are no studies published in PubMed, only in Scielo, Villalva-Rojas et al. [19,21] have reported a study on the bioequivalence of ibuprofen 400 mg tablets, concluding that they are bioequivalent [22]. While Herrera et al., reported a study on the in vitro therapeutic equivalence of three formulations of diazepam 10 mg tablets, finding that according to the f2 values, the generic C drugs are not equivalent, while the generic drugs A and B are equivalent [23]. In the year 2013, Alva et al. conducted an in vitro bioequivalence study of propranolol 40 mg tablets, concluding that multi-source tablets are not bioequivalent in vitro [24].
The objective of the present work was to determine the in vitro therapeutic equivalence of the 5 mg glibenclamide multi-source tablets with respect to the reference medicine; and as a statistical indicator of “equivalence” we used the similarity factor f2, the dissolution efficiency (EF%) and the mean dissolution time (MDT).
Materials And Method
Study samples
The samples studied were acquired in the drugstore of the city of Ica-Peru and of national origin, which consisted of:
• Multi-source drug: 200 tablets of generic 5 mg glibenclamide from a National Laboratory, assigned with the letter T for lot 10192186, RS NG-3390, expiration date 01-2019.
• Reference drug: 200 tablets of Glidiabet of 5 mg of Laboratory Ferrer, assigned with the letter R for the lot 1070625, RS N-12347, expiration date 07-2020.
Reagents
Hydrochloric acid 36% ACS was purchased from Merck Peruvian SA, sodium acetate ACS, potassium monobasic phosphate ACS, absolute ethanol anhydrous ACS, sodium hydroxide ACS were purchased from Mercantil Laboratory SAC [25].
A USP Sigma G2539-5G glibenclamide primary standard was used, which was purchased from Mercantil Laboratory SAC.
Materials and equipment
We used a Disolutor brand Electrolab TDT model of 8 stations, a UV/Vis 2550 Shimadzu Spectrophotometer and a Sartorius P22-1S Analytical Balance. All equipment used is calibrated and certified.
Method
An analytical method validated by López A et al., of ultraviolet absorption, at 300 nm was used [26].
Design of the in vitro therapeutic equivalence study: Preparation of the reference solution. Exactly 35 mg of standard glibenclamide was weighed and then transferred to a 50 ml flask, dissolved in 30 ml of sonicating ethanol for 15 minutes. It was brought to volume with the same solvent, obtaining a final concentration of 0.7 mg/ml, then eight dilutions were made with the following concentrations: 0.014, 0.028, 0.035, 0.042, 0.049, 0.070, 0.091 and 0.098 mg/ml of ethanol.
Then, the readings were made at a wavelength of 300 nm, using ethanol as target, obtaining the absorbance as a function of the concentration, to obtain the calibration curve with a R2 value of 0.993.26
Quantification of glibenclamide: The average weight of 20 tablets of glibenclamide 5 mg was determined and then they were pulverized. A quantity of powder equivalent to 3.5 mg of active ingredient was weighed, then transferred to a 50 ml flask with 30 ml of ethanol, and sonicated for 25 minutes. It was brought to volume with ethanol, obtaining a final concentration of 0.07 mg/ml immediately, we proceeded to filter with Acrodisc Syringe Filters Gelman Laboratory 0.45 μm, then the readings were made at a wavelength of 300 nm, using as ethanol target [26].
Determination of dissolution profiles. To perform the dissolution profile, we worked with 12 tablets for each formulation, generic glibenclamide 5 mg and Glidiabet 5 mg; USP type 2 device (pallet), study time 90 minutes, rotation speed 75 rpm, dissolution medium temperature 37 ± 0.5ºC, dissolution medium volume 900 ml, dissolution media 0.1 N hydrochloric acid pH 1.2, buffer acetate pH 4.5, phosphate buffer pH 6.8, aliquot taken with replacement of 5 ml. Pre-set sampling times 5, 10, 15, 30, 45, 60, 90 minutes. The samples were filtered with Acrodisc Syringe Filters Gelman Laboratory filter 0.45 μm and then the readings were made in duplicate at a wavelength of 300 nm, using the dissolution media as a target [26].
Analysis of data
As a statistical indicator of the “in vitro therapeutic equivalence” of the multi-source medicine with the referent, the similarity factor (f2) was used and calculated with the following equation: 5.19
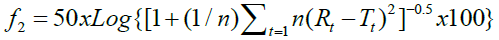
The efficiency of the solution (EF%) was calculated from the values obtained in the area under the curve (AUCot) of the dissolution profiles applying the trapezoids method and calculated with the following equation:
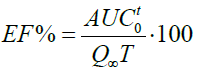
To characterize the dissolution profile of the drug, the mean dissolution time (MDT) was used and calculated with the following equation:
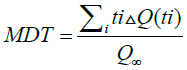
Results And Discussion
Figure 1 shows the average values of the dissolution profile of the generic glibenclamide compared to that referring to three pH. It can be seen that both drugs, at pH 1.2 and pH 4.5 do not dissolve at least 85% in 15 minutes, according to the Food and Drug Administration (FDA), or in 30 minutes, according to the Pharmacopoeia of the States United (USP); but at pH 6.8, both drugs meet this criterion. So that the similarity factor analysis (f2) is not applied, they must be dissolved in the three means of dissolution, according to the FDA; and according to the criteria of the USP, to consider them as fast-dissolving drugs and apply the bio exception, at least 85% of the drug must be dissolved in 30 minutes using the device 2 and to three dissolution media of different pH.25 Our results, confirm that the limiting factor for the absorption of glibenclamide, is its lack of dissolution due to its limited solubility, and that it could be one of the factors of therapeutic failures in clinical practice. Already in 2007, Pereda and Martínez, had demonstrated the deficiency of release of the drug, reporting that the drug referring Daonil dissolves in 50% at 30 minutes, and 32% nationally produced drugs, so that they did not meet the condition established by the FDA for in vitro equivalence studies [25]. In another study conducted by El-Sabawi et al., it was reported that the Daonil reference in Jordan and the generic drugs did not release a significant percentage of the drug in the first 30 minutes, in this case the Daonil showed the lowest percentage of release (20%), while the other products varied in a range of 35 to 65%. These studies were carried out in a medium of phosphate buffer solution at pH 6.8.19 Wei H and Löbenberg R., 2006, showed that the dissolution media have an impact on the solubility of glibenclamide [16]. Azharshekoufeh et al., demonstrated that the dissolution speed of glibenclamide is optimal with a combination of liquid-solid and co-grinding technologies [27,28]. Mor et al., have reported that the GLI: POL2 granule fusion provides a higher dissolution rate (85.11%) compared to other polymers [29]. Applying the ANOVA and the multiple comparison test of Tukey’s, results in the dissolution percentages of the multi-source drug T and the drug R for each of the times sampled at pH 1.2 and pH 4.5, present significant differences point to point for a probability of 0.05 (P<0.05). At pH 6.8 there is a significant difference from 5 to 30 minutes (P<0.05). Given that both criteria were met at pH 6.8, but not at pH 1.2 or at pH 4.5, these drugs produced in Peru cannot be considered rapid dissolution, so it is decided to apply the similarity factor.

Figure 1: Dissolution profile of the generic drug T and reference T at three pH (n=12).
Tables 1-3 show the results of the similarity factor (f2) of the multi-source tablets of 5 mg glibenclamide (T) compared to the reference (R). It is observed that the value of the factor f2, of the multi-source drug T at pH 1.2 is 76.60, and at pH 4.5 it is 72.12, which indicates a difference of less than 5%, while at pH 6, 8 is 82.49, the difference being less than 2% [30]. In this sense, the curves are similar and equivalent in vitro with the drug Glidiabet, which is sold in Peru. Unlike that reported by Pereda D and Martínez L., 2007, who when applying factor f2 determined that the generic glibenclamide medication is not equivalent, since the f2 was 41.1.26
| n | t (min) |
Dissolution profile at pH 1.2 | Similarity factor | |||||
|---|---|---|---|---|---|---|---|---|
| Referent drug R | Multi-source drug T | |||||||
| R (%D) |
CV (%) |
SD | T (%) |
CV (%) |
SD | f2 (50-100) | ||
| 1 | 5 | 27.07 | 2.00 | 0.542 | 26.05 | 3.54 | 0.921 | 76.60 |
| 2 | 10 | 39.50 | 2.02 | 0.800 | 38.30 | 2.70 | 1.033 | |
| 3 | 15 | 47.43 | 2.16 | 1.024 | 46.15 | 2.26 | 1.044 | |
| 4 | 30 | 64.27 | 2.28 | 1.468 | 68.15 | 1.44 | 0.982 | |
| 5 | 45 | 72.31 | 2.54 | 1.833 | 77.94 | 1.45 | 1.127 | |
| 6 | 60 | 78.63 | 3.62 | 2.849 | 78.35 | 1.45 | 1.138 | |
| 7 | 90 | 99.68 | 0.29 | 0.294 | 101.25 | 0.92 | 0.930 | |
Table 1: Similarity factor valúes f2 calcúlate with the data generated in Apparatus 2 USP from the solution of the multi-source drug and referring to pH 1.2.
| n | t (min) |
Dissolution profile at pH 4.5 | Similarity factor | |||||
|---|---|---|---|---|---|---|---|---|
| Referent drug R | Multi-source drug T | |||||||
| R (%D) |
CV (%) |
SD | T (%) |
CV (%) |
SD | f2 (50-100) | ||
| 1 | 5 | 20.11 | 2.39 | 0.479 | 16.11 | 4.86 | 0.783 | 72.12 |
| 2 | 10 | 31.83 | 2.28 | 0.724 | 26.58 | 4.97 | 1.320 | |
| 3 | 15 | 39.30 | 2.37 | 0.932 | 34.44 | 3.60 | 1.241 | |
| 4 | 30 | 52.41 | 3.01 | 1.579 | 50.40 | 2.61 | 1.315 | |
| 5 | 45 | 59.48 | 3.31 | 1.968 | 59.25 | 2.12 | 1.259 | |
| 6 | 60 | 64.81 | 3.39 | 2.199 | 65.62 | 2.07 | 1.357 | |
| 7 | 90 | 98.89 | 0.71 | 0.700 | 102.40 | 0.83 | 0.854 | |
Table 2: Similarity factor values f2 calculated with the data generated in Apparatus 2 USP from the solution of the multi-source drug and referring to pH 4.5.
| n | t (min) |
Dissolution profile at pH 6.8 | Similarity factor res | |||||
|---|---|---|---|---|---|---|---|---|
| Referent drug R | Multi-source drug T | |||||||
| R (%D) |
CV (%) |
SD | T (%) |
CV (%) |
SD | f2 (50-100) | ||
| 1 | 5 | 87.32 | 1.22 | 1.067 | 83.27 | 0.97 | 0.810 | 82.49 |
| 2 | 10 | 98.43 | 1.05 | 1.031 | 95.69 | 1.01 | 0.966 | |
| 3 | 15 | 100.15 | 0.81 | 0.809 | 99.70 | 0.92 | 0.918 | |
| 4 | 30 | 100.21 | 0.70 | 0.699 | 102.30 | 0.79 | 0.809 | |
| 5 | 45 | 100.37 | 0.64 | 0.639 | 100.71 | 0.85 | 0.856 | |
| 6 | 60 | 100.30 | 0.61 | 0.609 | 100.13 | 0.98 | 0.984 | |
| 7 | 90 | 100.34 | 0.81 | 0.816 | 100.72 | 0.61 | 0.616 | |
Table 3: Values of similarity factor f2 calculated with the data generated in Apparatus 2 USP from the solution of the multi-source drug and referring to pH 6.8.
Table 4 shows the values of the independent model parameters that characterize the drug release curve for glibenclamide drugs of 5 mg in the three study pH. In the case of dissolution efficiency (EF%), this parameter measures the area under the drug dissolution curve over a period of 90 minutes, and indicates the degree of absorption in vivo and can be theoretically related to the concentration curve plasma activity as a function of the time obtained by deconvolution techniques of in vivo data [28], exhibiting a value of 95.98% for the multi-source drug T, compared to the referent that exhibited a EF of 96.35%, both at a pH of 6.8. While the values of EF% for the multi-source T and reference R to pH 1.2 are 68.66 and 68.22%, and at pH 4.5 it is 56.59 and 59.24%, respectively; In this regard, the resulting value is greater than 50% for immediate-release medications, which is why it shows in vitro equivalence.
| Study drug | pH 1,2 | pH 4,5 | pH 6,8 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| AUCoT (min%) |
EF (%) |
MDT (min) |
AUCoT (min%) |
EF (%) |
MDT (min) |
AUCoT (min%) |
EF (%) |
MDT (min) |
|
| Referent drug R | 306.0 | 68.2 | 28.5 | 263.6 | 59.2 | 36.3 | 435.04 | 96.4 | 3.34 |
| Multi-source drug T | 312.8 | 68.7 | 28.6 | 260.8 | 56.6 | 39.9 | 435.03 | 95.9 | 4.54 |
Table 4: Values of the independent model parameters at three pH.
The mean dissolution time (MDT) in vitro was calculated in order to characterize the rate of dissolution of the drug. In pharmaceutical forms of immediate release, it indicates the average time required for the dissolution of the drug, being in our study of 4.54 minutes for the multi-source T, and for the reference of 3.34 minutes at a pH of 6.8; while at pH 1.2 both drugs T and R, have a dissolution speed of 28 minutes, and at pH 4.5, their speed is between 56.59 to 59.24 minutes, which correlates with the profile of dissolution. This parameter is very relevant, as mentioned by Mady O., since it is used to establish an in vitro and in vivo correlation [13], due to the fact that the average gastric residence time (T50%) is 15-20 minutes. under fasting conditions. If the solution is slower than gastric emptying, a dissolution profile with multiple time points in multiple media is recommended, and in any case carry out relative bioavailability studies to demonstrate the bioequivalence and interchange ability of the multi-source drug.
Conclusions
The present study reveals significant differences in the dissolution profiles at two pH, but can be considered similar to the reference drug R according to the similarity factor (f2) and bio pharmaceutically equivalent depending on the efficiency of dissolution (EF%). However, it is recommended to carry out properly controlled relative bioavailability studies to demonstrate if they are bioequivalent and interchangeable.
Sources of Financing
This article has been funded by the authors.
Conflicts of Interest
The authors declare no conflict of interest.
References
- http://www.who.int/ medicines/ services/ expertcommittees/pharmprep/QAS04_093Rev4_final.pdf
- Alvarado A, Salazar A, Pineda N, Villanueva H, Cáceres E (2016) Study of the relative bioavailability of a multi-source sulfamethoxazole formulation with respect to the reference medicine. Horiz Med 16: 12-19.
- Nainar S, Rajiah K, Angamuthu S, Prabakaran D, Kasibhatta R (2012) Biopharmaceutical Classification System in in vitro/ in vivo Correlation: Concept and Development Strategies in Drug Delivery. Tropical J Pharmaceutical Res 11: 319-329.
- Mahesh K, Anil B (2012) Biopharmaceutics drug disposition classification system: An extensión of Biopharmaceutics Classification System. Inter Res J Pharm 3: 5-10.
- Jung Cook H, De Anda G, Rubio K, Mayet L (2012) Comparación de perfiles de disolución. Impacto de los criterios de diferentes agencias regulatorias en el cálculo de f2. Rev Mex Cienc Farm 43: 67-71.
- Medina J, Hurtado M, Cortés A, DomÃnguez A (2012) Disolución comparativa de indometacina en cápsulas utilizando los Aparatos 1 y 4 USP. Rev Mex Cienc Farm 43: 72-80.
- Saavedra I, Iturriaga, Ãvila L, Quiñones L (2011) Estudios de bioexención (in vitro) para establecer equivalencia de medicamentos. Cuad Med Soc 51: 66-79.
- Tabbakhian M, Hasanzadeh F, Tavakoli N, Jamshidian Z (2014) Dissolution enhancement of glibenclamide by solid dispersion: solvent evaporation versus a supercritical fluid-based solvent-antisolvent technique. Res Pharm Sci 9: 337-350.
- Stavchansky S (2008) Scientific Perspectives on Extending the Provision for Waivers of In vivo Bioavailability and Bioequivalence Studies for Drug Products Containing High Solubility-Low Permeability Drugs (BCS-Class 3). The AAPS J 10: 300-305.
- León G, Osorio M, Matiz G (2015) Estudio biofarmacéutico comparativo de tabletas de acetaminofén 500 mg disponibles en el mercado colombiano. Rev Cubana Farma 49: 630-640.
- Boniatti J, Pereira A, Carnevale de Souza A, Rodrigues C, Assis da Costa M, et al. (2014) Galenic approaches in troubleshooting of glibenclamide tablet adhesion in compression machine punches. Saudi Pharm J 22: 445-453.
- Mady O (2014) Studying the effect of dispersed drug crystal in the organic phase on the encapsulation by solvent evaporation technique (3) Independent models as tools for studying the drug release profiles. World J Pharm Sci 2: 409-421.
- Villarroel A, Clement Y, Sealy P, Löbenberg R, Montane-Jaime L, et al. (2015) Comparing the Dissolution Profiles of Seven Metformin Formulations in Simulated Intestinal Fluid. Dissolut Technologie 2015: 17-21.
- Singh S, Srinivasan K, Gowthamarajan K, Narayan G (2010) Development and validation of discriminatory dissolution procedure for poorly soluble glyburide. Asian J Pharmac 2010: 205-212.
- Wei H, Löbenberg R (2006) Biorelevant dissolution media as a predictive tool for glyburide a class II drug. Eur J Pharm Sci 29: 45-52.
- Alemón R, Chávez J, RamÃrez B, Rivera L, GarcÃa R (2014) Cuantificación sanguÃnea de metformina y glibenclamida para endocrinologÃa pediátrica. Acta Pediátr Mex 35: 373-380.
- Zharikova O, Ravindran S, Nanovskaya T, Hill R, Hankins G, et al. (2007) Kinetics of glyburide metabolism by hepatic and placental microsomes of human and baboon. Biochemical Pharmacology 73: 2012-2019.
- El-Sabawi D, Abbasi S, Alja'fari S, Hamdan I (2013) Pharmaceutical evaluation of glibenclamide products available in the Jordanian market. Afr J Pharm Pharmacol 7: 1464-1470.
- Dos Santos E, Pires R, Lara-Filho M, Saraiva A, Fregonezi M (2007) Dissolution Test for glibenclamida tablets. Quim Nova 30: 1218-1221.
- RodrÃguez M, Luna J, Carlucci A, Bregni C (2004) Relación de Estabilidad Termodinámica Relativa entre Polimorfos de Glibenclamida. Acta Farm Bonaerense 23: 169-175.
- Villalva-Rojas O, Grande-Ortiz M, Ortiz J, Isasi J, Yantas D, et al. (2007) Estudio de bioequivalencia del ibuprofeno 400 mg tabletas. Rev Peru Med Exp Salud Publica 24: 356-362.
- Herrera-Calderon O, Grande-Ortiz M (2012) Equivalencia terapéutica de tabletas de diazepam dispensadas en la ciudad de Ica. Perú Rev Med Hered 23: 154-159.
- Alva P, RuidÃas D, Quiliche J, Sánchez Y (2013) Bioequivalencia in vitro de tabletas de propranolol 40 mg multifuente e innovador. Revista Pharmaciencia 1: 28-34.
- Pereda D, MartÃnez L (2007) Daonil y glibenclamida 5 mg de producción nacional: liberación in vitro. Rev Cubana Farm 41: 1-7.
- López M, Felizola A, Hernández V, Cuartero T (2005) Validación de un método analÃtico espectrofotométrico para cuantificar glibenclamida en tabletas de 5 mg. Rev Mex Cienc Farm 36: 33-41.
- Pérez M, Orobio Y, Baena Y (2013) Estudio comparativo de la liberación in vitro de metformina a partir de dos productos multifuente de liberación inmediata, comercializado en Colombia. Re Colomb Cienc QuÃm Farm 42: 169-189.
- Azharshekoufeh L, Shokri J, Barzegar-Jalali M, Javadzadeh Y (2017) Liquigroud technique: a new concept for enhancing dissolution rate of glibenclamide by combination of liquisolid and co-grinding technologies. BioImpacts 7: 5-12.
- Mor J, Batra D, Ahlawat S (2016) Enhancement of drug dissolution of glibenclamida using solid dispersion technique. The Pharma Innovation J 5: 8-12.
- Franco-Ospina L, Matiz-Melo G, Pájaro-BolÃvar I (2012) Estudio biofarmacéutico comparativo de marcas comerciales de tabletas de ciprofloxacino disponibles en el mercado colombiano. Rev Salud Pública 14: 695-709.
Citation: Alvarado-Yarasca A, Muñoz-Jauregui AM, Quiñones-Sepúlveda L, Lizaraso-Soto F, Salazar-Granara A, et al. (2017) Study of in vitro Therapeutic Equivalence of the 5 mg Glibenclamide Multi-source Tablets Respecting the Reference Medicine Product. Clin Pharmacol Biopharm 6:180. DOI: 10.4172/2167-065X.1000180
Copyright: © 2017 Alvarado-Yarasca A, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Share This Article
Recommended Journals
Open Access Journals
Article Tools
Article Usage
- Total views: 5573
- [From(publication date): 0-2017 - Dec 21, 2024]
- Breakdown by view type
- HTML page views: 4810
- PDF downloads: 763