Paired-End Phylogenetic Analysis of Bacterial Population Differences in Mus Musculus Cecum in Response to Synbiotic Administration
Received: 15-Dec-2023 / Manuscript No. JIDT-23-122913 / Editor assigned: 18-Dec-2023 / PreQC No. JIDT-23-122913 (PQ) / Reviewed: 02-Jan-2024 / QC No. JIDT-23-122913 / Revised: 09-Jan-2024 / Manuscript No. JIDT-23-122913 (R) / Published Date: 16-Jan-2024 DOI: 10.4172/2332-0877.1000577
Abstract
Background: The cecum has the highest metabolite absorption and houses the most abundant population of microbes which are responsible for maintaining homeostasis and host health. Disruption of the microbial populations in either presence or relative abundance due to various environmental and host factors can result in various disease state that do not have a standard form of medical treatment.
Materials and methods: The use of synbiotics, prebiotics, and probiotics has shown potential in treating dysbiosis. Many gut microbiota cannot be isolated by culture-dependent techniques. Culture independent, high resolution paired-end16S rRNA genomic analysis of the V3/V4 hypervariable region was used to elucidate bacterial differences under treatment.
Results: Analysis shows the establishment of probiotic strains maintaining diversity of the gut microbial flora within the cecum under the additional supplementation of prebiotics.
Conclusion: This study demonstrated a measurable phylogenetic difference in microbial community composition among controlled and symbiotic diets supporting the use of probiotics in establishing homeostatic balance of beneficial bacteria and pharmaceutically correcting a state of dysbiosis in the host.
Keywords: Paired-end; Prebiotic; Probiotic; Synbiotic; Diversity; 16S rRNA; Next-gen sequencing
Keywords
Paired-end; Prebiotic; Probiotic; Synbiotic; Diversity; 16S rRNA; Next-gen sequencing
Introduction
Microbial communities abound in a range of environments including that of the human host which contains upwards of 100 trillion microbial cells, outnumbering human eukaryotic cells tenfold. The most densely populated environment is located in the lower intestine of the gastrointestinal tract, where the human gut microbiota and its genome are known collectively as the ‘gut microbiome.’ A variety of exogenous metabolic and genetic features bestowed from microbe to host are the result of a co-evolved, symbiotic relationship between these two entities [1-7]. For example, the gut microbiome is largely responsible for the mediation of host health through means of moderating host homeostasis, development and regulation of the immune response, contributing to pathogen colonization resistance, and actively participating in nutrition and metabolism [5,6,8-11]. Epithelial development, regulation of energy homeostasis, blood circulation, and adaptive and innate immunity mechanisms are also influenced by the host gut microbiome. Disruptions in the normal activity of host interactions of this microbial community including adaptations to the various aspects of the ‘modern lifestyle’ including travel, diet, age, geographic locale, stress, and use of medications can therefore result in a diseased state [1,12-30].
A number of interventions have been integrated into the management of the dysbiotic state in the gut microbiome as there is currently no standard or agreed-upon method for treatment due to the exhibited broad range of effectiveness. Such methodologies include antibiotic administration, bacteriotherapy, and fecal-oral transplantation [1,7,15,25,29,31,32]. Antibiotics are generally administered due to this lack of effective treatment and are often unsuccessful, resulting in recurrent infections and altering the microbial community composition at a rate of roughly 90% [20,32]. Promising, yet highly invasive and requiring an extensive screening process, fecal- oral transplantations are also employed but with varying success rates [1,31-34]. Utilization of the emerging pre-, pro-, post-, and synbiotics has shown increasing potential for correcting the dysbiotic state and maintaining host health [1,7,12-14,16,18-27,29,30,35-37].
Probiotics are classified as “organisms and substances which contribute to intestinal microbial balance” or “a live microbial feed supplement which beneficially affects the host animal by improving its intestinal microbial balance” and must meet specific guidelines by remaining stable and viable under storage and use, be prepared on a large scale, and benefit host health after introduction in addition to surviving and remaining present in the integrated ecosystem after the product used for initial introduction has been depleted [1,20,26,36]. Commonly used probiotics that show effectiveness in various pathologies include Lactobacilli (Lactobacillus acidophilus, L. casei, L.delbruekii) Bifidobacteria (Bifidobacterium adolescentis, B. bifidum, B. longum, B. infatis), and Streptococci (Streptococcus salivarius, S. thermophiles, S. lactis) [20,37,38]. However, L. acidophilus is the most prevalently utilized and tolerated probiotic, and it has been shown to synthesize vitamin K which is necessary for the conversion of the bone matrix osteocalcin to the active form and may thus aid in improving bone integrity [1,12-14,18-27,29,30,37,38].
Prebiotics are “a non-digestible food ingredient that beneficially affects the host by selectively stimulating the growth and/or activity of one or a limited number of bacteria in the colon, and thus improves host health [1].” Like probiotics, additional criteria must be satisfied. A prebiotic cannot be absorbed or hydrolyzed in the upper GI tract, and it must serve selectively as a substrate for specific commensal, beneficial bacteria for growth or metabolic activation. It also must alter the microbial composition to that of a healthy state, and cause systemic or luminal effects that benefit host health [1,20,26,36]. Compounds meeting these specifications include non-digestible foods such as oligosaccharides, polysaccharides, fructooligosaccharides, and other naturally occurring non-digestible carbohydrates, peptides, proteins, and lipids. Host health is improved through the stimulation of growth and activity of specific endogenous microbiota by altering the microbial composition in a given locale due to supplementation of nutrients and metabolites [20,36].
The term ‘synbiotics’ includes the combined use pro- and prebiotics and is defined as “a mixture of probiotics and prebiotics that beneficially affects the host by improving the survival and implantation of live microbial dietary supplements in the gastrointestinal tract, by selectively stimulating the growth and/or by activating the metabolism of one or a limited number of health-promoting bacteria, and thus improving host welfare [1].” Synbiotics have been shown to combat the diseased dysbiosis state via supplementation with functional and health enhancing nutrition by maintenance of the colonic flora in the healthy, balanced state. The combined usage of pre- and probiotics has been shown to increase the effectiveness of each individual component [1,20,26,31,36,37].
The 16S rRNA amplicons commonly utilized in community profiles and diversity can aid in the detection of rare phylotypes, ecological characteristics, and taxonomic identification due to it being present in all organisms, being large enough for informatics purposes (at roughly 1500 bp) while containing both conserved (the most highly conserved structural element in rRNA) and hypervariable loop regions, not being transferred horizontally, and being considered universal- thus greatly increased the rise of complex, novel microbial consortia [39-42].
The V3/V4 loop region of the bacterial 16S ribosome can be analyzed through utilization of paired-end sequencing and annotation [43]. The paired-end analysis technique is advantageous to other high-throughput sequencing techniques in that it reduces the amount of erroneous sequences that are included in downstream analysis (imposing a quality control step) while providing enormous data sets.
The detection of microbial flora and metabolites directly resulting from administration of probiotic supplementation compared to the controlled population in this study provides optimal insight regarding the alleviation and eradication of the disease states caused by an imbalance or absence of the aforementioned.
Materials and Methods
Sample preparation
The harvested cecum from male 9-month old mice (Harlan), Mus musculus following either a control diet (B,C,F) or experimental (symbiotic) (H,J,L) diet were chosen at random. The diets, based on a powdered form of American Institute of Nutrition (AIN)-93M purified rat diet (Dyets, Inc., Bethlehem, PA), were administered to the mouse model and were modified to utilize cornstarch in place of sucrose and dextrin in order to reduce the susceptibility of osmotic dehydration of the bacteria studied within the synbiotic diet. These isocaloric diets were developed according to carbohydrate ingredient manipulation by assuming energy densities of 4, 0, and 2 kcal/g for cornstarch, cellulose, and fructooligosaccharide, respectively. The synbiotic diet contained prebiotics in the form of fructooligosaccharides and probiotic cultures (Nutraceutix, Redmond, WA) composed of 1 × 1011 CFU/g of equal parts Lactobacillus acidophilus and Lactococcus lactis. The diets were made fresh three times a week with addition of probiotics immediately prior to feeding each morning for 18 weeks [44].
The triplicate diet-specific cecum samples were sterilely dissected both laterally and vertically and rehydrated using 6 mL 10 mM TRIS, pH~8.0, 1% Triton after being stored at -20˚C. Samples were then incubated at 80˚C for one hour, and centrifuged (1000 rpm) for five minutes to achieve a pellet. Residual supernatant was removed after centrifugation at 16,000x g for 10 minutes. The cell pellet obtained was resuspended in 250 uL 10 mM TRIS.
DNA isolation
Genomic DNA of the cecum samples was obtained by a mechanical sheer forces protocol utilizing 100 μL Lysozyme (100 mg/mL) added to the rehydrated cell suspension solution and incubated for 30 min at 37˚C. After which, 100 μL Proteinase K (10 mg/mL in 10 mM TRIS) was added, incubated for 30 min, and brought up to volume with lysis buffer (500 mM NaCl, 50 mM EDTA, 1% SDS (w/v), and 50 mM TRIS (pH 8.0)) in a bead-beating tube. Prior to supernatant being drawn off and added to isopropanol, tubes were then bead-beat for 5 min, boiled at 80˚C for 10 min, microcentrifuged at 3000x G for 5 minutes, and stored at -20˚C for 12 h. Samples were then microcentrifuged at 13000xG for 10 min, and the pellet washed with 200 μL 70% ethanol (4˚C) prior to centrifugation at 13000xG for 10 min. Ethanol was removed by drying at 37˚C for 60 min before DNA was rehydrated in 10 mM TRIS, pH ~8.0.
Paired-end and PCR amplification
A gradient bacterial paired-end PCR (200 μM/L 16S paired-end bacterial designed primers [forward primer: 5’-TCG TCG GCA GCG TCA GAT GTG TAT AAG AGA CAG CCT ACG GGN GGC WGC AG-3’ and reverse primer: 5’-GTC TCG TGG GCT CGG AGA TGT GTA TAA GAG ACA GGA CTA CHV GGG TAT CTA ATC C-3’] , Vent polymerase, 40-60˚C annealing temperature with a four minute extension time) was run on an Escherichia coli K12 (+) control which yielded an optimized temperature of 41.5˚C for annealing. A 16S PCR was then run on the genomic DNA samples in triplicate in 50 μL reactions for each of the genomic DNA samples with each reaction containing the following: The same 16S paired-end primers (Integrated DNA Technologies), Vent polymerase (New England Biolabs, Ipswich, MA), 1x ThermoPol Buffer (New England Biolabs, Ipsich, MA), 400 μM per each deoxynucleotide triphosphate (New England Biolabs, Ipswich, MA), and 1 μL genomic DNA template. Individual master mixes and negative controls were used for each sample in a program consisting of the following steps: 10 min denaturation at 95˚C, followed by 30 cycles of 95˚C/1 min, optimized annealing for 2 min at 41.5˚C, and 4 min at 72˚C. The final cycle of the previously listed steps was immediately followed by a concluding 10 min elongation step at 72˚C. After a low EEO 1% agarose “check” gel verified the results, the best of each of the samples (dictated by present DNA concentration as verified by Image J software) was Nano Dropped (NanoDrop ND1000 Spectrophotometer, Thermo Scientific, Wilmington, DE) in triplicate to achieve a proper DNA concentration, and submitted to Idaho State University Molecular Research Core Facility (MCRF) for flow cell sequencing using the Illumina 16S Metagenomic Sequencing Library Protocol (Illumina), the Illumina Nextera XT Index Kit for PCR indexing, and the Illumina MiSeq Reagent Kit v.3 600 cycle chip and the MiSeq Software Suite for sequencing on the Illumina MiSeq 2 Instrument allowing automated generation of DNA clonal clusters via bridge amplification and analysis through utilization of a reversible dye terminator.
Annotation and phylogenetic analysis
Mothur: Paired sequence files were processed using the Galaxy platform (https://galaxyproject.org/) and annotated according to the Mothur platform (https://www.mothur.org) using the 1.33.3 MiSeq SOP default settings (https://www.mothur.org/wiki/MiSeq_SOP) with the following exceptions: Sequences shorter than 35 nt and longer than 600 nt (with a limit set at 500) having homopolymers longer than 8 nt were omitted from further analysis in addition to reads with ambiguous base calls or incorrect primer sequences, a ‘pre.cluster’ command was utilized to denoise and identify OTUs by applying a pseudo-single linkage algorithm to remove sequences subjected to pyrosequencing errors, putative chimeras were identified and removed utilizing the Chimera Uchime algorithm, ‘pcr.seqs’ command was run across the entire SILVA v.4 database, clustering was accomplished via the ‘cluster. split’ command, taxonomic classification of each identified OTU was established by setting the distance matrix cutoff at .2 to avoid clustering below 80% similarity and a species cut off set at .03, and a final cut off was set at .1 for genus level identification at 95% and species at 97% according to the RDP (Ribosomal Database Project) database [45-48].
Normalized average and standard deviations of taxonomic abundance values of significant phylogenetic identifications were obtained from the final file produced by the Mothur of 16S paired- end sequencing of control and synbiotic diet samples. An increase or decrease in abundance was determined from synbiotic values relative to their control counterpart to show the effects of the synbiotic treatment on the diversity and phylogenetic profile of the gut microbial flora (Table 1). Significant values are shown in Figure 1, where an increase in abundance in synbiotic samples comparative to control samples is depicted by a closed diamond, and decrease an open diamond.
A 1000 boot strap value maximum likelihood phylogenetic tree having a 50% majority rule of significance was then generated in Bio Edit software from collected genus level strain assignments produced by Mothur analysis of the 16S paired-end genomic DNA of the diet specific mouse cecum samples (Figure 1). Significant strain determined according to a T-test algorithm (data not shown) with all singletons, sequences having a threshold occurrence value less than 10, sequence lengths less than 1400 base or greater than 2300 bases of 16S paired-end sequencing according to type strains present on the Ribosomal Data Base (http://rdp.cme.msu.edu) removed were then truncated on the V3/V4 loop region of the ribosome. Thermus aquaticus was used as the outgroup.
MG-RAST: Joined paired-end sequence files were uploaded to MG-RAST (https://www.mg-rast.org/), phylogenetic trees restricted to bacteria having maximum levels of order, genus, and species were generated in addition to analyses of rarefaction, PCOA, and heatmaps. Protein coding and ribosomal gene prediction in MG-RAST was accomplished utilizing the protein FragGeneScan and ribosomal gene BLAT similarity searches. This in turn in was utilized to depict the alpha diversity, or description of species, within a given sample. BLAT was also utilized to identify homologous sequences in the M5NR database. Alignments were made according to sequence similarity comparisons, and amino acid alignments utilizing FragGeneScan to collect ORF predictions prior to BLAT integration for translated amino acid sequence identification within the M5NR database. Annotation within MG-RAST were based on putative gene function collected from the public databases previously discussed [49].
Results
Control diet cecum composition comparisons
Analysis via MG-RAST of control diet sample ‘B,’ uploaded on 9/24/14, produced a total of 567 identified ribosomal RNAs (from the predicted 758 predicted rRNA features) from 24,579 (4.7%) of the 521,139 sequences (205,384,411 bps) that passed quality control filtering algorithms (pre-quality control bp count at 231,268,701 bp). 608 sequences failed and were removed from downstream analysis. The uploaded mean sequence length was 443 ± 14bp and uploaded mean GC content 54 ± 2% which compares to 394 ± 3% mean sequence length post quality control and GC percent post quality control of 54 ± 3%. SILVA SSU database generated the highest identification of annotated ribosomal RNA genes (24418), followed by RDP (20096), Greengenes (19671), and SILVA LSU (1) all of which had an e-value raised to -30 and less. F15,945,780 sequences are present in the M5NR protein database which include all unique sequences from applied protein databases, and 309,342 sequences in the M5RNA ribosomal database that contains unique sequences from the utilized ribosomal RNA databases. 97.1% of the detected sequences were of the bacterial domain.
A total number of 316,307 sequences containing 189,417,901 bp, and having an average length of 589 ± 7 bp was initially uploaded for MG-RAST analysis of control diet sample ‘H’ on 9/30/2014. 444 sequences, or 0.1%, failed to pass the imposed quality control pipeline imposed in this software. Post quality control mean sequence length was 599 bp and GC % 56 ± 3%. Of those sequences that passed quality control, only 10,766 sequences (3.4%) contained ribosomal RNA genes with 719 alignment identified rRNA features from the initially predicted 1,205. The number of features identifies were annotated by the datasets employed in this sample analysis including protein databases, protein databases containing functional hierarchy information, and rRNA databases. Different databases yield varying results due to the completeness of annotated data contained within the database. SILVA SSU database generated the highest identification of annotated ribosomal RNA genes (9758), followed by RDP (7394), and Greengenes (7183) with an e-value raised to -30 and less. 88.4% of detected sequences belonged to the bacterial domain.
MG-RAST analysis of control diet mouse cecum sample J (9/30/2014) resulted in a total number of 774,847 sequences totaling 463,639,316 bp with an average length of 598 bp ± 13 bp and a mean GC content of 56 ± 3%. 0.3% of the sequences (2,307 sequences) failed to pass the quality control. The post quality control sequences (772,540 and 462,750,694 bp) had a mean sequence length of 598 ± 1 bp and a mean GC content of 56 ± 3%. 35,277 sequences (4.6%) of the sequences that passed quality control contained rRNA genes with 1,194 rRNA features identified from the predicted 2,992. SILVA SSU database generated the highest identification of annotated ribosomal RNA genes (30887), followed by RDP (27684), Greengenes (23560), and SILVA LSU (1) with an e-value raised to -30 and less. 98.1% of the detected sequences belonged to bacteria.
Phylum level taxonomic hits distribution denotes the most abundant phyla in mouse cecum sample B as the following: Firmicutes, 30217 (68.3%); Verrucomicrobia, 4990 (11.3%); Actinobacteria, 3674 (8.3%); Proteobacteria, 2321 (5.2%); Tenericutes, 795; Unclassified (derived from bacteria), 474; Bacteroidetes, 327; Chordata, 73; Cyanobacteria, 69; Synergistetes, 62; and Unclassified sequences, 57; where listed abundance of annotations were derived on a log scale and is representative of richness and evenness of that taxonomic level within the given sample. Phylum level assignments in sample H also lead with Firmicutes being the most abundant (13704 at 64.4%) while Actinobacteria (3799, 17.8%) were the following most prevalent assignments. Both samples contained ‘main players’ composed of many the same phylum but at different relative abundances (sample H also contained: Tenericutes, 460; Cyanobacteria, 302; Proteobacteria, 289; Bacteroidetes, 91; Unclassified (derived from bacteria), 77; Verrucomicrobia, 75; and unclassified (derived from other sequences), 41). Sample J also followed the same patterns previously expressed and was composed of the following ‘main players’ on a phylum level: Firmicutes 64.3%, 33263; Verrucomicrobia 9.8%, 5071; Actinobacteria 9.3%, 4834; Bacteroidetes 9.2%; 4775; Proteobacteria, 1270; Tenericutes, 756; Unclassified (derived from Bacteria), 463; Cyanobacteria, 292; and Unclassified (derived from unclassified sequences), 246 (data not shown) (Figures S1-S20).
Class level taxonomic abundance between control diet mouse cecum samples B, H, and J also were composed of many the same assignments. The most prevalent class amongst the group was Clostridia (B: 26421, 59.7%; H: 7059, 33.2%; and J: 22672, 43.8%). Class assignments of Clostridia dominated the most in both B and J while H was closely followed by Bacilli at 28.9% (6150). Bacilli was high in the other two samples but not as such comparative proportions (8512 or 16.4% in sample J and 3283 or 7.4% in B). Actinobacteria (3674 [8.3%], 3799 [17.8%], 4834 [9.3]) and Mollicutes (795, 460, 756) were also relatively abundant in class assignments for the control diet B, H and J samples, respectively (Figures S7 and S9).
Taxonomic abundance according to order level distinctions among the control mice B, H, and J was led by the most abundant Clostridiales (26421, 59.7%; 7056, 33.1%; 22667, 43.8%). Other predominant order identifications included Lactobacillales, Bifidobacteriales, Bacillales, and Actinomycetales (data not shown).
The lack of grouping exhibited by the three control diet samples in PCOA analysis demonstrates dissimilarity of taxonomic or functional abundance profiles amongst the control diet samples. The reduced dimensionality of this graphic depiction to limited variables simplifies the data contained within these samples. Neither axis demonstrates a high r2 value for the dataset (Figure S13). Raw abundance counts relative to each control diet sample is illustrated in Figure S15 while normalized values for the control samples is depicted in Figure S17. Distribution of these abundance counts is denoted as a positive integer between 0 and 1 (a uniform scaling that has no impact on the value differences within a single sample or between samples) for the number of times a particular taxon has been detected is explicative of comparisons of normality of the relative distributions of abundance. As raw abundance value distribution characteristically varies among samples (Figure S15), there is still enough similarity amongst the samples as is evident by the clustering at the bottom of each of the samples. The normalization of the values (Figure S17) by a common variable allowing for data of different scales to be compared and generated for the samples, reduced the variation among the sample distributions of abundance for a more concise analysis of the five number summary (the minimum, first quartile or 25% coverage, median, third quartile or 75% coverage, and maximum) of abundance values for each sample. Each of the samples had a median line at roughly 0.3, a first quartile of ~0.1, and a third quartile nearing 0.6. Some variation was evident in the minimum and maximum values, as J had the lowest, nearing zero, followed by B, and H had the highest maximum value being the closest to 1. J had the lowest maximum value.
Rarefaction curves generated for control diet fed mouse cecum samples reflected annotated species richness where the annotation number total was derived as a function of the original number of sampled sequences. Species abundance is reflected by the initial curve evident in Figures S1-S3 for the control samples as the data generating the curve is calculated by the observed abundance of species within the utilized datasets subsample annotations. Each of the figures demonstrated a sharp initial slope indicative of novel species within the initiation of the analysis. The rounding off or eventual plateau observed from decreased slope steepness with increasing number of reads correlates to adequate sampling within each of the control samples where additional sampling is unlikely to result in a significant amount of detected novel species assignments. The general trend observed within this graphic analysis is a sharp initial rise followed by a plateau at an asymptote as demonstrative of decreasing detection of new species per unit of collected individuals. Figure 2 depicts this trend by superimposing all rarefaction curves for samples of the control diet. J had the largest slope, as is represented its higher alpha diversity value followed by B and H. However, sample H did have a larger alpha diversity than B but also contained less reads than B.
The alpha diversity, or relative organism diversity with a single number in a sample, of control diet cecum sample B is 36.412 species within the range of two standard deviations and is estimated from species level annotation distribution which is a measure of species richness. Shannon diversity is reflected by the average weighted abundance of the logarithmic value of relative abundance of the generated annotated species within this sample as collected by annotation source databases utilized in MG-RAST. Mouse B had the lowest observed alpha diversity among the control diet cecum samples followed sample H (44.185 species) and the highest value of 55.725 species in sample J (data not shown).
K-mer profile of the k-mer rank abundance of 15-mer coverage according to sequence size for sample B yielded a decrease of optimal coverage of 1202604 after 67 sequences from the initial generated plateau until 1.2 e^ 6 sequences when no detectable coverage was measured (data not shown). This correlates to a decrease in abundance of high-coverage, repetitive sequences as analysis progressed within this sample set. In comparison, sample H yielded a decrease of optimal coverage of 442413 after 25 sequences from the initial generated plateau until 8.89 e^ 6 sequences when no detectable coverage was measured. Sample J also produced a decrease of optimal coverage from a value of 1202604 after the pattern exhibited by mouse H [50].
Synbiotic diet cecum composition comparisons
A total number of 664,839 sequences containing 398,224,212 bp, and having an average length of 598 ± 5 bp was initially uploaded for MG-RAST analysis of synbiotic diet sample ‘C’ on 9/26/2014. 417 sequences, or 0.1%, failed to pass the imposed quality control pipeline. Post quality control mean sequence length was 599 bp and GC % 55 ± 3%. Of those sequences that passed quality control, only 20,139 sequences (3.0%) contained ribosomal RNA genes with 1,145 alignment identified rRNA features from the initially predicted 2,502. SILVA SSU database generated the highest identification of annotated ribosomal RNA genes (17820), followed by RDP (17710), and Greengenes (16170) with an e-value raised to -30 and less. 94.6% of detected sequences belonged to the bacterial domain.
MG-RAST analysis of synbiotic sample ‘F’ on 9/30/14 contained 1,079,526 sequences having 646,611,472 bp and an average length of 598 ± 5 bp from which 650 sequences, or 0.1% failed quality control filtering pipelines within the software package. 66,505 (6.2%) of the post quality control sequences which had a mean sequence length of 599 bp and GC% of 54 ± 3% contained ribosomal RNA genes with 2,291 of the predicted 4,322 aligned rRNA features being identified. SILVA SSU database generated the most identified ribosomal RNA genes (61126) with RDP (60122), Greengenes (59102), and SILVA LSU (1) following. 98.5% of the detected sequences were of the bacterial domain.
Analysis of the synbiotic diet sample ‘L’ on 9/30/2014 was performed on 926,558 sequences totaling 554,892,258 bp having an average length of 598 ± 6 bp. 721 sequences (0.1%) failed to pass the quality control. Post quality control sequences had a mean sequence length of 599 bp and a mean GC percent content of 56 ± 3% with 2,335 rRNA features identified of the initially predicted 3,573. Source hits distribution was highest with the SILVA SSU database (24964) followed by RDP (23587), and Greengenes (22592) databases. 96.4% of the detected sequences were of the bacterial domain.
Phylum level taxonomic hits distribution for individual synbiotic diet cecum samples result in the most abundant classification, measuring at 74.6% (30249), of the phyla assignments generated for synbiotic mouse C belonged to Firmicutes. Actinobacteria (11.1%, 4488), Verrucomicrobia (1665), Proteobacteria (748), Tenericutes (366), and Cyanobacteria (420) then followed as the most abundant identifications for this sample. Samples F and L also resulted with many of the same phyla being the most prevalent, respectively: Firmicutes (69.6%, 67777; 66.1%, 37812), Actinobacteria (13.2%, 12891; 22.9%, 13106), Verrucomicrobia (12.3%, 11981), Proteobacteria, (1669, 1151), Cyanobacteria (567, 863), and Tenericutes (410,1765). Many of the following phyla assignments also correlated, just at differing abundances per sample (data not shown).
Class level taxonomic abundance between synbiotic diet mouse cecum samples C, F, and L also were composed of many the same assignments. All lead with Bacilli (37.6%, 15263; 46.7%, 45447; 41.3%, 23625) and had many of the same following class assignments at relatively high abundance including: Clostridia (29.8%, 12103; 21.9%, 21328; 17.55%, 9993), Actinobacteria (29.8%, 4488; 13.2%, 12891; 17.5%; 13106), Erysipelotrichi (7.1%, 2866; 299; 7%, 4008), Verrucomicrobiae (1665; 12.3%, 11981; 79), and Gammaproteobacteria (540; 848; 675) (Figures S8 and S10).
Order level distinctions of taxonomic abundance among the synbiotic mice C,F, and L were led by Lactobacillales (30.5%, 12374; 41.5%, 40376; 34.4%, 19702) and followed by Clostridiales (29.8%, 12093; 21.9%, 21324; 17.5%, 9986), Bacillales (7.1%, 2889; 5.2%, 5071; 6.9%, 3923), and Bifidobacteriales (6.5%, 2639; 9.3%, 9018; 18.2%, 10390) as the most prevalent distinctions. Comparative to assignments based on the level of class, order distinctions are less in agreement across the three samples of the synbiotic diet samples (data not shown).
PCOA analysis of the synbiotic samples (Figure S14) demonstrates reduced dimensionality and dissimilarity of taxonomic or functional abundance profiles through the lack of clustering in the position of the samples on the Figure. Comparative to the results of the control data previously discussed, neither axis contained a high r2 value for the dataset (PCO1, 0.61386; PCO2, 0.38614). Raw abundance counts relative to each control diet sample is illustrated in Figure S16 while normalized values for the control samples is depicted in Figure S18. Distribution of these abundance counts depicts normality of the taxon detection amount as explained previously. There was variation among the raw abundance count distribution despite the similarity exhibited among the samples (Figure S16). Each sample had two data points distributed higher than the rest of the data and mean numbers which were clustered at the bottom of the graphic. However, sample F, listed as the first sample on the Figure, had higher placement of the top two data points comparative to the other two samples. The remaining data points were similar across samples. Normalized values (Figure S18) generated a median line similar to the control diet at roughly 0.3. However, the first quartile of the synbiotic diet samples was higher at roughly 0.2, but the third quartile was similar to the control diet by being located at roughly 0.6 for each of the samples. Although all of the samples had a minimum value less than 1, C was the highest, and F was the lowest. C also produced the highest maximum value of the diet samples, nearing 1, while both F and L were closer at roughly 0.9.
Synbiotic diet fed mouse cecum samples rarefaction curves (Figures S4-S6 for samples C, F, and L, respectively) depicting the species richness of the annotations within the samples is shown by the initial steep with high relative slope and curve produced in each of the Figures. Each the curves produced a plateau with increasing read number to show adequate sampling were detection of novel species assignments would be unlikely with integration of additional samples in the analysis. Figure 3 depicts this trend by superimposing all rarefaction curves for samples of the synbiotic diet. F had the largest slope and most reads followed by C and L. However, sample L contained more reads than that of sample C.
Measured alpha diversity also differed amongst the samples. Although it had the largest slope, sample F had the lowest alpha diversity of the synbiotic diet at a value of 25.40 species, while C had the highest generated alpha diversity measurement at 43.36 followed by L at 38.17.
K-mer rank abundance of 15-mer coverage according to sequence size yielded a profile in sample C depicting a decrease of optimal coverage of 1202604 right before the plateau formed by the initial 25 sequences until 8.89e^6 sequences when no detectable coverage was measured (data not shown). The level of coverage of rare sequences followed by high-coverage, repetitive sequences is shown by this decreasing value across sequences. Sample F yielded a decrease of optimal coverage of 3269017 after 25 (further than sample C) sequences from the initial generated plateau until 2.2 e^ 7 sequences when no detectable coverage was measured. Sample L also produced a decrease of optimal coverage at the same value of C but at the same point in sequencing as F.
Cecum composition comparison between diets
The normalized average of taxonomic abundance values of significant phylogenetic identifications of control and synbiotic diet samples showed an increase in strain abundance in all taxons listed in Table 1 with the exception of Nocardioides genus, Clostridia class, Clostridiales order, and Lachnospiraceae family while comparing synbiotic treatment results to control diet results. The measured increase in abundance was determined from synbiotic sample values relative to the control (Table 1). Not all significant genus level assignment abundances were included in the Mothur-based tree. Included within the table were higher level taxonomic classifications which showed significant abundances that may not have included the genus level assignments such as Sphingomonas or higher levels of taxonomic classification beyond the designation included in the figure.
A 1000 bootstrap 16S paired-end genus level phylogenetic rectangular consensus cladogram according to a 50% majority rule of significant singletons as derived from a T-test (data not shown) and containing sequences with a less than 10 threshold occurrence value, length between 1400 and 2300 bases was generated from data produced by the Mothur software package for type strains present in the Ribosomal Data Base (Figure 1). Firmicutes and Actinobacteria were among the most prevalent phylogenetic assignments, followed by Gammaproteobacteria, Alphaproteobacteria, Bacteriodetes, and Betaproteobacteria, increased statistical significance is integrated by use of 100 bootstraps for resampling. The shorter the branch lengths, the increased relation among the designations, as is evident among the proteobacteria. Both of the supplemented probiotic genus were detected and are present among the Firmicutes. Other commonly utilized strains of probiotics, as previously discussed, are also present. As this figure is representative of the type strains present in RDP, exact phylogenetic identity is unknown. However, this figure is representative of the immense diversity present within the cecum. Figure 1 is limited compared to the phylogenetic analyses generated by MG-RAST in that assignments are not depicted according to individual sample or diet but collectively according to the integration of all samples from both analyzed diets. The Mothur-based tree contained more strain abundance increases when comparing synbiotic to the control diet, however not all designations included on the table appeared on the figure.

Figure 1: Maximum likelihood 1000 boot strap value phylogenetic rectangular consensus cladogram with 50% majority rule of significant (according to a T-test algorithm [data not shown] with all singletons, sequences having a threshold occurrence value less than 10, sequence lengths less than 1400 base or greater than 2300 bases removed) genus level strains reported by Mothur analysis of 16S paired-end sequencing according to type strains present on the Ribosomal Data Base. Significant taxa increase from normalized averages as shown in Table 1 is depicted by a closed diamond and decrease by an open diamond.
Comparative MG-RAST analysis of all of the samples from both the control diet mice B (MG-RAST accession 4581535.3), J (4582209.3), and H (4582210.3) and synbiotic C (MG-RAST accession 4581845.3), F (4582170.3), and L (4582211.3) diet was performed on 11/19/2014 and finished on 2/10/2015. The 16S paired-end phylogenetic trees of the cecum microbiome of the samples yielded the following color designations for the samples as is depicted in the center of the Figure: B, silver; C, blue; F, red; H, gold; J, purple; and L, green (Figures 4, S19, S20). Data for each of the Figures was compared to M5NR according to a maximum e-value of 1e-5, 60% minimum identity, and an alignment minimum length of 15 (amino acid measurement according to protein and bp in RNA databases). Each of the figures also contained stacked bar leaf weights but differed in the maximum phylogenetic level and taxonomic coloring assignments (Figure S19 contained a genus maximum and class coloring, Figure S20 species maximum and class coloring, and Figure 4 species maximum and order coloring). Figure S19 showed the highest abundance of class level identification among Clostridia followed by Gammaproteobacteria, Bacilli, and Actinobacteria. Many of the samples amongst both diets contained the same genus level identifications. It is notable that all of the strains contained the Lactobacillus probiotic which was added to the synbiotic diet. Unlike Figure 1, Lactococcus was not detected in this figure. Bifidobacterium, a common probiotic utilized, was also present in all of the samples. Figure S20 provides species level classification in concordance to the trends depicted in Figure S19. However, the supplemented probiotics utilized in the synbiotic diet were not detected through analysis utilizing MG-RAST. Many of the commonly utilized strains of probiotics are present, however, the most prevalent order, as depicted by Figure 4, is that of Clostridiales followed by Bacillilales and Lactobacillilaes-all of which are present in each of the 6 analyzed samples. Analysis of these figures shows that there is not a designated prevalence of genus or species according to diet, as assignments are not limited to the three samples composing the diet, but include at least one sample of the opposing diet. There are phylogenetic identifications listed that are specific to a given sample, however, as is depicted by the singular bar coloring found at the center of the figure.

Figure 2: Rarefaction curve of 16S bacterial paired-end fragments of cecum microbiome of control diet mice, B, F, and J generated by MG-RAST. Data was compared to M5NR
according to a maximum e-value of 1e-5, 60% minimum identity, and an alignment minimum length of 15 (amino acid measurement according to protein and bp in RNA databases).
The top, blue line is representative of sample J, having an alpha diversity value of 55.72; middle, orange line is representative of sample B, having an alpha diversity value of 36.41;
and bottom, red line mouse H, having an alpha diversity of 44.18 (data not shown). 
A normalized bar chart comparing the detected bacteria according to groups designated by diet with corresponding p-values is provided in Figure 5. The data was compared to M5NR with a 1 e-5 e value maximum, minimum identity at 60%, and minimum alignment length of 15 bp according to RNA database hits. The smallest p value, which indicates the most distinct difference between the diet groups, was found in Actinobacteria with a value of 0.104, and the highest p value in Spirochaetes with a value of 1. Probiotic-containing Firmicutes was the most abundant and had a p value of 0.1165, followed by Actinobacteria and Cyanobacteria (0.1628).

Figure 3: Rarefaction curve of 16S bacterial paired-end fragments of cecum microbiome of synbiotic diet mice, C, F, and L. Data was compared to M5NR according to a maximum
e-value of 1e-5, 60% minimum identity, and an alignment minimum length of 15 (amino acid measurement according to protein and bp in RNA databases). The top, orange line is
representative of sample F, having an alpha diversity value of 25.40; middle, blue line is representative of sample C, having an alpha diversity value of 43.36; and bottom, red line
mouse L, having an alpha diversity of 38.17 (data not shown). 
Discussion
The exact identity of species shifts between diseased and healthy homeostatic states of host health has remained elusive, although limited information regarding phylum-level changes has been obtained. This impedes understanding of microbial community interactions during treatments of dysbiosis due to the heterogeneity of the GI tract as a result of differences within microenvironments and spatial distribution of flora and metabolites. This can cause variations of effectiveness and activity of pre- and probiotics by location within the gut microbiota as the metabolic products of one bacterium can be modified and utilized by another bacterial species. Increasing the availability of a molecule in its active form can be enhanced by community level biotransformation reactions. These cooperative interactions directly affect the degree of effectiveness of a prebiotic, as the necessary active form may never reach its target location, or a probiotic, which may contain a strain that does not yield the desired, beneficial effect on microbial composition and function [5,6,20,37] .
OTU assignments were utilized in this study according to the derived genetic distance between sequences. Distribution of sequence abundances among OTUs allowed for general estimates of ecological richness, evenness, and diversity of the community in addition to measurements of the like between communities of differing diet supplementation. Phylogenetic methods employed investigated differences in communities according to sequence difference. The application of the OTU approach allowed for quantitative measurements to be collected [51]. However, it is noted that employing the OTU definition can result in an overestimation in similarity amongst community comparisons [52]. Past research has claimed that accurate distance-based threshold for taxonomic level definitions can be created through consensus-based methods of OTU classification. This is due to the inability to define bacterial taxonomic levels resulting from the lack of adequate bacterial taxa being cultured or culturable [51]. Many of the present taxonomic outlines and requirements are based on previously cultured organisms causing candidate phyla and non-culturable phyla that are lacking in taxonomy identifying to the level of genus or species. However, there is currently no accepted and employed definition of a bacterial species which increases the difficulty in appropriate taxonomic classification according to phylotype or even the genera, family, class, order, or phyla of bacteria. The operational definition for a species cites a 3% dissimilarity, but it is not widely accepted. Also, pre-clustering followed by clustering at 3% (equivalent to 97% sequence identity which shows shifts in clusters of higher and reduced dominance) as employed in this study allowed for increased accuracy in OTU characterization in addition to providing a reduction of singleton sequence proportion and minimally affecting the distribution and presence of microbial taxa. Single nucleotide errors also had minimal effect on classification of sequences. Methods employing OTU approaches avoid many of the limitations implicit to phylotype analysis due to the lack of bin restrictions since taxonomy outlines are not applied. Sequences can thus be assignment and clustered with equal basis regardless of reference sequence representation or restrictions issued in outline classifications. OTU assignment is depended on the presence of other sequences in the dataset. However, this methodology assumes that the 16S bacterial rRNA gene evolves at the same rate among all taxonomic affiliations which is disputed [28,53].
Despite the varying sequences produced in each of the samples within the two diets, sequences had similar length (roughly 600 bp) and contained comparable GC % content (between 50 and 60%) after the quality control pipelines imposed during analysis via MG-RAST. SILVA and RDP databases produced the most hits among all samples. Phylogenetic analysis results produced by the extensive filtering and quality control pipelines employed by both Mothur and MG-RAST were comparable but varied to some degree. Data produced from Mothur analysis were later graphically represented based on type strains collected on the Ribosomal Data Base of statistically relevant sequences based on OTU assignments due to the computational, memory, and time limitations imposed by the Mothur software-a methodology not applied to the same sequencing files imputed into MG-RAST, as previously discussed [54]. It is also due to these limitations that individual phylogenetic assignments could not be designated to the separate samples or between the diets studied. Mothur identified Firmicutes and Actinobacteria as the most prevalent phylogenetic assignments, followed by Gammaproteobacteria, Alphaproteobacteria, Bacteriodetes, and Betaproteobacteria. Also, shorter branch lengths and therefore increased relationship between designations were most evident among Proteobacteria. Other commonly utilized strains of probiotics were also present on the Mothur-generated phylogenetic analysis of identity among all of the samples. However, as Mothur- generated results were representative of the type strains present in RDP, exact identity is unknown, limiting the data compared to the phylogenetic analyses generated by MG-RAST in that assignments are not depicted according to individual sample or diet but collectively according to the integration of all samples from both analyzed diets. Yet, immense diversity present within the cecum is still apparent from the results generated.
The normalized average of taxonomic abundance values of significant phylogenetic identifications of control and synbiotic diet samples showed an increase in strain abundance in all taxa listed in Table 1 with the exception of Nocardioides genus, Clostridia class, Clostridiales order, and Lachnospiraceae family when comparing synbiotic sample values relative to the control (Table 1). The indigenous gut flora determines the incidence of C. difficile colonization and infection. Clostridia have shown to induce various degrees of dysbiosis on the gut microbiome, detrimentally affecting host heath as previously discussed. A reduction of abundance, but not complete removal of presence, conserves microbial diversity within the gut microbial community while decreasing the propensity of disease development. Nocardioides has not shown any involvement in human pathology. Dominant colonization with Lachnospiraceae bacterial family is common in the diseased mammalian host, and has shown to have an inverse relationship with Clostridia. The significant increase in abundance of the strains included in the probiotic supplementation within the synbiotic diet contribute to the effectiveness of the probiotic by achieving the requirements previously discussed. Not all significant genus level assignment abundances were included in the Mothur- based tree. Included within the table were higher level taxonomic classifications which showed significant abundances that may not have included the genus level assignments such as Sphingomonas or higher levels of taxonomic classification beyond the designation included in the figure.
| Taxanomic Level | Taxon | Control Average | Control Standard Deviation | Synbiotic Average | Synbiotic Standard Deviation | Increase | Decrease |
|---|---|---|---|---|---|---|---|
| 6 | Unclassified Acidobacteria | 0 | 0 | 1.3008618 | 2.253158738 | * | |
| 6 | Unclassified Acidimicrobiales | 0 | 0 | 0.5259785 | 0.911021435 | * | |
| 6 | Kocuria | 0 | 0 | 0.9837642 | 0.975747665 | * | |
| 5 | Nocardiaceae | 0 | 0 | 1.0519569 | 1.822042869 | * | |
| 6 | Rhodococcus | 0 | 0 | 1.0519569 | 1.822042869 | * | |
| 6 | Aeromicrobium | 0 | 0 | 0.3333333 | 0.577350269 | * | |
| 6 | Nocardioides | 1.474009324 | 1.609876591 | 0 | 0 | * | |
| 4 | Nitriliruptorales | 0 | 0 | 1.8268403 | 1.963162868 | * | |
| 5 | Nitriliruptoraceae | 0 | 0 | 1.8268403 | 1.963162868 | * | |
| 6 | Nitriliruptor | 0 | 0 | 1.8268403 | 1.963162868 | * | |
| 4 | Solirubrobacterales | 0 | 0 | 1.1764094 | 1.035762123 | * | |
| 5 | Conexibacteraceae | 0 | 0 | 1.1764094 | 1.035762123 | * | |
| 6 | Conexibacter | 0 | 0 | 1.1764094 | 1.035762123 | * | |
| 5 | Prevotellaceae | 0 | 0 | 1.1764094 | 1.035762123 | * | |
| 6 | Prevotella | 0 | 0 | 1.1764094 | 1.035762123 | * | |
| 5 | Sphingobacteriaceae | 0 | 0 | 0.6666667 | 1.154700538 | * | |
| 6 | Sphingobacterium | 0 | 0 | 0.6666667 | 1.154700538 | * | |
| 6 | Chelatococcus | 0 | 0 | 1.1764094 | 1.035762123 | * | |
| 6 | Agromonas | 0 | 0 | 0.3333333 | 0.577350269 | * | |
| 4 | Sphingomonadales | 3.43529961 | 0.249407594 | 6.0389708 | 1.917194353 | * | |
| 5 | Sphingomonadaceae | 3.43529961 | 0.249407594 | 5.3885399 | 1.224108691 | * | |
| 6 | Acinetobacter | 0 | 0 | 0.6666667 | 1.154700538 | * | |
| 6 | Pseudomonas | 0.980645143 | 0.882758007 | 3.2283663 | 1.405243312 | * | |
| 6 | Moraxella | 0 | 0 | 0.5259785 | 0.911021435 | * | |
| 3 | Bacilli | 7693.249296 | 4193.816257 | 22078.153 | 2393.512187 | * | |
| 4 | Lactobacillales | 7666.847041 | 4177.209249 | 22034.675 | 2372.19706 | * | |
| 5 | Aerococcaceae | 0 | 0 | 0.6666667 | 1.154700538 | * | |
| 6 | Aerococcus | 0 | 0 | 0.6666667 | 1.154700538 | * | |
| 5 | Enterococcaceae | 3.85089896 | 5.257604443 | 20.687218 | 12.64405135 | * | |
| 6 | Enterococcus | 3.85089896 | 5.257604443 | 19.703453 | 11.66832969 | * | |
| 5 | Lactobacillaceae | 7326.48491 | 4182.193139 | 13659.472 | 785.1739086 | * | |
| 6 | Lactobacillus | 6926.249723 | 4327.384756 | 13085.724 | 792.5078663 | * | |
| 5 | Streptococcaceae | 156.6942946 | 57.45560938 | 8014.3968 | 2483.549268 | * | |
| 6 | Lactococcus | 17.5148527 | 6.451849537 | 7943.218 | 2490.62685 | * | |
| 6 | unclassified | 2.294081843 | 2.002301437 | 12.21863 | 2.983466826 | * | |
| 5 | unclassified | 172.436285 | 85.70878406 | 328.55115 | 78.23330943 | * | |
| 6 | unclassified | 172.436285 | 85.70878406 | 328.55115 | 78.23330943 | * | |
| 3 | Clostridia | 24984.25364 | 4590.072698 | 10322.344 | 2991.153003 | * | |
| 4 | Clostridiales | 24956.43237 | 4591.263597 | 10299.223 | 2992.641808 | * | |
| 5 | Eubacteriaceae | 11.61933553 | 2.677567758 | 35.002209 | 4.604352731 | * | |
| 6 | Anaerofustis | 10.63869039 | 3.291712254 | 34.668876 | 4.53162293 | * | |
| 5 | Lachnospiraceae | 17512.64269 | 5257.16041 | 7585.2706 | 2141.076544 | * | |
| 6 | Moryella | 0 | 0 | 1.0519569 | 1.822042869 | * | |
| 6 | Faecalibacterium | 0 | 0 | 0.6666667 | 1.154700538 | * |
Note: *: Increase and Decrease in synbiotic values
Table 1: Normalized average taxonomic abundance values from Mothur analysis of 16S paired-end sequencing of control and synbiotic diet samples. An increase or decrease in abundance is determined from synbiotic values relative to their control counterpart.
Application of the Mothur data can be made to the results generated to MG-RAST as comparative analysis of all of the samples from both the control and synbiotic diets (as capable of the MG-RAST software) (Figures 4, S19, and S20) showed the highest abundance of class level identification among Clostridia followed by Gammaproteobacteria, Bacilli, Actinobacteria, similar genus level identifications, and the presence of probiotic Lactobacillus which was added to the synbiotic diet. Unlike Mothur results, Lactococcus was not detected. However, the common probiotic Bifidobacterium was identified in all of the samples. The supplemented probiotics utilized in the synbiotic diet were not detected through analysis utilizing MG-RAST by species level classifications; yet, like Mothur results, many of the commonly utilized strains of probiotics were identified. The most abundant order in all of the samples was identified as Clostridiales followed by Bacillilales and Lactobacillilaes. However, individual analysis of these figures shows that there is not a designated prevalence of genus or species according to diet, as assignments are not limited to the three samples composing the diet, but include at least one sample of the opposing diet. The smallest p value indicating the most distinct difference between the diet groups was found in Actinobacteria with a value of 0.104, and the highest p value in Spirochaetes with a value of 1. Probiotic-containing Firmicutes was the most abundant and had a p value of 0.1165, followed by Actinobacteria and Cyanobacteria (0.1628) (Figure 5). The distance between branches within the trees correlate to the relation of the identifications included.

Figure 4: 16S paired-end bacterial phylogenetic tree of cecum microbiome of control diet mice B (MG-RAST accession 4581535.3), J (4582209.3), and H (4582210.3) and synbiotic
samples C (4581845.3), F (4582170.3), and L (4582211.3) having stacked bar leaf weights, a species maximum level, and coloring according to order. Data was compared to
M5NR according to a maximum e-value of 1e-5, 60% minimum identity, and an alignment minimum length of 15 (amino acid measurement according to protein and bp in RNA databases). Order membership was indicated by the color of the species names. 
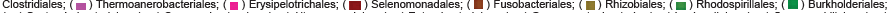


Figure 5: 6A normalized bar chart of control diet samples.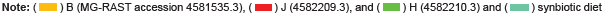
 in brackets. M5NR was compared to the data with e-value maximum of 1e-5, 60% minimum identity, minimum alignment length of 15bp in RNA databases.
in brackets. M5NR was compared to the data with e-value maximum of 1e-5, 60% minimum identity, minimum alignment length of 15bp in RNA databases.
Individual MG-RAST mitigated analyses of both the control and synbiotic diet samples identified Firmicutes as containing the highest number of phylum level taxonomic hits with the same phyla assignments following in abundance though not always in the same order among samples. Class level taxonomic abundance between control diet mouse cecum samples also were composed of many the same assignments as Clostridia dominated and less prevalent assignments were among the same groupings of Class. Despite class level taxonomic abundance between synbiotic diet mouse cecum samples containing of many the same assignments within that diet, the most abundant was not the same as the control diet samples as all lead with Bacilli. Abundance according to order level distinctions within the control yielded Clostridiales as the most abundant compared to synbiotic samples which identified Lactobacillales as the most abundant. This is notable as it correlates to the supplemented strain of probiotic within the diet, and supports the effectiveness of the synbiotic treatment combination as successfully establishing and selectively cultivating the probiotic strain through the presence of the prebiotic compounds contained in that diet (data not shown).
PCOA analysis of the control and synbiotic samples demonstrates reduced dimensionality and dissimilarity of taxonomic or functional abundance profiles through the lack of clustering in the position of the samples (Figures S13 and S14). Comparative to the results of the control data, neither axis contained a high r2 value for the dataset. The distribution of raw abundance and normalized depicts normality of the taxon detection amount as previously mentioned. Variation among the raw abundance count distribution was evident in both diets due to clustering patterns despite the similarity exhibited among the samples. Normalized values generated a median line at 0.3 for both diets. However, the first quartile of the synbiotic diet samples was higher at roughly 0.2 compared to the control diet at ~0.1, and the third quartile location was roughly 0.6 for both diets. Although all of the samples had a minimum value less than 0.1, there was variation amongst the samples of each of the diets. This trend was also evident in analysis of the maximum value, where all sample locations were less than 1, but varied to some degree among diets. Transformation of data to achieve a normal Gaussian distribution via Normalization resulted in the reduction of variation or biases introduced throughout sample preparation and analysis that are not under experimental control in addition to decreasing their impact on interpretation of results (Figures S17 and S18). This also improve the results exhibited in other comparative analyses (PCA, heatmap, etc) that assume a normal distribution of data for applicable, statistically relevant data interpretation. Means of data normalization are discussed in further detail on the MG-RAST V 3.0 database. The comparable results evident between the diets is represented of the stability of the gut microbiomes in both of the diets. This could represent a general homeostatic balance required for functionality in a healthy host. The variations observed correlate to the individual differences in the profiles analyzed which could be attributed to the difference in taxonomic or functional abundances produced by the supplementation of probiotic strains in the synbiotic diet as previously discussed.
Rarefaction curves reflecting annotated species richness where the annotation number total was derived as a function of the original number of sampled sequences produced an initial steep slope representing species for each of the diets. This sharp initial slope indicates of novel species within the initiation of the analysis of both of the samples. Both diets also yielded an eventual plateau at an asymptote from decreased slope steepness with increasing number of reads correlating to adequate sampling among diets where additional sampling is unlikely to result in a significant amount of detected novel species assignments. Each of the samples within each diet produced variation in the steepness of the initial slope with varying read amounts. The slope of the samples did not correlate to alpha diversity levels among diets as is evident in the variation among samples. The control diet generated a higher average alpha diversity among samples at 44.441 compared to the synbiotics 35.643 species. This could be due to the difference in the number of available reads between samples, however as this measure is dependent on that variable.
The k-mer rank abundance profile of 15-mer coverage according to sequence size yielded a decrease of optimal coverage at less than 70 sequences for all samples contained in both diets from the initial generated plateau until no detectable coverage was measured (data not shown). Redundancy, or repetitiveness, of sequences within each sample according to occurrence of distinct 15 bp patterns is representative of rare, or low coverage sequences being initially depicted, followed by high-coverage, repetitive sequences (the decrease on coverage previously discussed). The rank abundance plot is a function of abundance rank with the most abundant sequences being first listed. This represents a decrease in abundance of high-coverage, repetitive sequences as analysis progressed within this sample set. Use of k-mer applications allows for the identification of the closest sequence template for the generated data pertaining to a given sample. These results could represent taxonomic diversity increasing in lower dominant distributions [47,50,54,55].
As technology advances, the challenge with phylogenetic analysis methodology has shifted from sample sequence generation to sequence analysis. MG-RAST serves as a high-throughput pipeline for high performance computing and annotation allowing a low cost, next generation means of worldwide metagenomic sequence analysis. The functional sequence assignments of the metagenomic input produced by this software package are automated and generated from both nucleotide and protein database comparison allowing for functional summaries and comparative phylogenetic analysis [47,53]. Biological databases are growing exponentially, and algorithms that minimize processor time and memory requirements are becoming increasingly important, which was problematic while utilizing Mothur. Analysis and clustering algorithms are also computationally intensive for OTU- based methodology. Mothur utilizes a neighbor joining algorithm that is taxonomy-independent and performs better than most deterministic and heuristic methods available. However, the generated OTUs can represent sequences from multiple lineage assignments due to there being no taxonomic level threshold commonly employed. The genetic distance between full 16S gene sequences in a given taxonomic assignment were continuous in each hierarchy level [51].
Even prior to the establishment of next generation sequencing as a primary method of analysis, the 16S rRNA gene was the most represented gene present in the GenBank database. However, various biases such as a microbial population relative abundance misrepresentation in a given sample and errors including actual sequence misrepresentation as a result of PCR sequencing and amplification can be present when utilizing the 16S rRNA gene in sequence survey. The DNA extraction method, DNA purification protocol, selection of PCR primers, PCR cycling conditions (PCR polymerases erroneously result in substitution in 1 of 10^5 to 10^6 bases, and amplification of heterogeneous templates can result in formation of chimeras (not a sequencing error) when incomplete amplification products are present to serve as primers for related fragments at a rate of 5-45%. Sequencing also results in errors due to the homopolymer under-representation at a rate of 0.01-0.02, community composition within the sample, and copy number of the 16S gene within the genome can affect whether the relative abundances of the gene being sequenced are equal to the bacterial presence in the sample. These biases confound the representation and application of data collected. Additionally, microbial ecology analyses include the inherent hypothesis that microbial community structure changes directly affect the function within the community [28,56].
Most studies assume that partial sequence distances are not significantly different from full-length sequence distances despite the fact that the 16S rRNA gene lacks uniform evolution throughout its length. When employing the proxy species definition specific to full length sequences 3% distance cut off, the variability in evolution within the 16S rRNA gene becomes apparent. Genetic diversity also decreases along the 16S rRNA gene length. Regression coefficients do not adequately explain variation between regions in comparison to the whole gene. Longer reads increase the relation of segmented analysis to the whole gene. The 16S gene is a marker for diversity within a genome and follows a well-determined secondary structure. The analysis of this gene via next gen sequencing allows for replicates to be analyzed in addition to increased complexity of experimental designs to be investigated while increasing the breadth and depth of sampling. Technical limitations are based on conserved PCR primer availability, fragment length, and gene quality generation while analytical limitations are dependent on accurate sequence classification and genetic diversity within a region availability. This necessitates the use of only a select region of a gene to be studied. Differing regions will be selected until analysis becomes standardized [52]. However, the datasets that are currently available for comparison are not completely exhaustive as there has been shown to be as few as 10% or as high as 98% lack of sequence matching from a sample to a dataset. The accuracy of annotations is dependent on the quality of the data used [49]. This also applies to beta-diversity studies, though useful in community comparisons, have limited use to only communities exhibiting clear differences and does not provide information pertaining to the details of these differences due to database-dependent methods that are limited according to the lack of representation of rare and novel populations when analyzing the deep coverage existent in many environmental samples [46,57-71].
Conclusion
The results generated in this study indicate there was a measurable phylogenetic difference in microbial community composition between the mouse diet groups, thus supporting the use of synbiotics as an effective means of establishing homeostatic balance of beneficial bacteria within the cecum content of the host. This also supports beneficial applications in regards to pharmaceutical intervention for correcting a dysbiotic state in the gut microbial flora of a diseased host over the alternative, invasive and possible harmful choices of intervention. However, it is noted that competition among microbes for nutrients and ecological sites as well as stress can also cause a decrease in effectiveness in this treatment. Further research is needed to investigate and characterize the intestinal communities of microbes, their metabolic activity, and functionality to increase the efficiency and effectiveness of synbiotic treatments.
References
- Cammarota G, Ianiro G, Bibbὸ S, Gasbarrini A (2014) Gut microbiota modulation: probiotics, antibiotics or fecal microbiota transplantation? Int Emer Med 9: 365-373.
[Crossref] [Google Scholar] [PubMed]
- Dethlefsen L, McFall-Ngai M, Relman DA (2007) An ecological and evolutionary perspective on human-microbe mutalism and disease. Nature 449: 811-818.
[Crossref] [Google Scholar] [PubMed]
- Druart C, Neyrinck AM, Vlaeminck B, Fievez V, Cani PD, et al. (2014) Role of the lower and upper intestine in the production and absorption of gut microbiota-derived pufa metabolites. PLoS ONE 9: e87560.
[Crossref] [Google Scholar] [PubMed]
- Ley RE, Peterson DA, Gordon JI (2006) Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell 124: 837-848.
[Crossref] [Google Scholar] [PubMed]
- Shi N, Li N, Duan X, Niu H (2017) Interaction between the gut microbiome and mucosal immune system. Military Med Res 4:14.
[Crossref] [Google Scholar] [PubMed]
- Singh RK, Chang HW, Yan D, Lee K, Ucmak D, et al. (2017) Influence of diet on the gut microbiome and implications for human health. J Transl Med 15: 73.
[Crossref] [Google Scholar] [PubMed]
- Walsh CJ, Guinane CM, O’Toole PW, Cotter PD (2004) Beneficial modulation of the gut microbiota. FEBS lett 588: 4120-4130.
[Crossref] [Google Scholar] [PubMed]
- Hooper LV (2004) Bacterial contributions to mammalian gut development. Trends Microbiol 12: 129-134.
[Crossref] [Google Scholar] [PubMed]
- Li M, Wang B, Zhang M, Rantalainen M, Wang S, et al. (2008) Symbiotic gut microbes modulate human metabolic phenotypes. Proc Natl Acad Sci USA 105: 2117-2122.
[Crossref] [Google Scholar] [PubMed]
- Mazmanian SK, Liu CH, Tzianabos AO, Kasper DL (2005) An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell 122:107-118.
[Crossref] [Google Scholar] [PubMed]
- Rankoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R (2004) Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 118: 229-241.
[Crossref] [Google Scholar] [PubMed]
- de Wit N, Derrien M, Bosch-Vermeulen H, Oosterink E, Keshtkar S, et al. (2012) Saturated fat stimulates obesity and hepatic steatosis and affects gut microbiota composition by an enhanced overflow of dietary fat to the distal intestine. Am J Physiol Gastrointest Liver Physiol 303: G589-599.
[Crossref] [Google Scholar] [PubMed]
- Cotter PD (2011) Small intestine microbiota. Curr Opin Gastroenterol 27: 99-105.
- Curtis TP, Sloan WT (2004) Prokaryotic diversity and its limits: microbial community structure in nature and implications for microbial ecology. Ecol Ind Microbiol 7: 221-226.
[Crossref] [Google Scholar] [PubMed]
- Dethlefsen L, Huse S, Sogin ML, Relman DA (2008) The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol 6: 2383-2400.
[Crossref] [Google Scholar] [PubMed]
- Druart C, Neyrinck AM, Dewulf EM, de Backer FC, Possemiers S, et al. (2013) Implication of fermentable carbohydrates targeting the gut microbiota on conjugated linoleic acid production in high-fat-fed mice. Br J Nutr 1-14.
[Crossref] [Google Scholar] [PubMed]
- Frit JV, Desai MS, Shah P, Schneider JG, Wilmes P (2013) From meta-omics to causality: experimental models for human microbiome research. Microbiome 1:14.
[Crossref] [Google Scholar] [PubMed]
- Gi Langille M, Meehan CJ, Koenig JE, Dhanani AS, Rose RA, et al. (2014) Microbial shifts in the aging mouse gut. Microbiome 2: 50.
[Crossref] [Google Scholar] [PubMed]
- Hayashi H, Takahashi R, Nishi T, Sakamoto M, Benno Y (2005) Molecular analysis of jejunal, ileal, caecal and recto-sigmoidal human colonic microbiota using 16S rRNA gene libraries and terminal restriction fragment length polymorphism. J Med Microbiol 54: 1093-1101.
[Crossref] [Google Scholar] [PubMed]
- Klemashevich C, Wu C, Howsmon D, Alaniz RC, Lee K, et al. (2014) Rational identification of diet-derived postbiotics for improving intestinal microbiota function. Curr Opin Biotechnol 26: 85-90.
[Crossref] [Google Scholar] [PubMed]
- Mujico JR, Baccan GC, Gheorghe A, Diaz LE, Marcos A (2013) Changes in gut microbiota due to supplemented fatty acids in diet-induced obese mice. Br J Nutr 1-10.
[Crossref] [Google Scholar] [PubMed]
- Niot I, Poirier H, Tran TT, Besnard P (2009) Intestinal absorption of long-chain fatty acids: evidence and uncertainties. Prog Lipid Res 48:101-111.
[Crossref] [Google Scholar] [PubMed]
- O’Hara AM, Shanahan F (2006) The gut flora as a forgotten organ. EMBO Rep 7:688-693.
[Crossref] [Google Scholar] [PubMed]
- O’Shea EF, Cotter PD, Stanton C, Ross RP, Hill C (2012) Production of bioactive substances by intestinal bacteria as a basis for explaining probiotic mechanisms: bacteriocins and conjugated linoleic acid. Int J Food Microbiol 152: 189-205.
[Crossref] [Google Scholar] [PubMed]
- Saulnier DM, Ringel Y, Heyman MB, Foster JA, Bercik P, et al. (2013) The intestinal microbiome, probiotics and prebiotics in neurogastroenterology. Gut Microbes 1: 17-27.
[Crossref] [Google Scholar] [PubMed]
- Saulnier DM, Santos F, Roos S, Mistretta T, Spinler JK, et al. (2011) Exploring metabolic pathway reconstruction and genome-wide expression profiling in Lactobacillus reuteri to define functional probiotic profiles. PLoS One 6 :e18783.
[Crossref] [Google Scholar] [PubMed]
- Sartor RB (2008) Microbial influences in inflammatory bowel diseases. Gastroenterol 134:577-594.
[Crossref] [Google Scholar] [PubMed]
- Schloss PD, Gevers D, Westcott SL (2011) Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS ONE 6: e27310.
[Crossref] [Google Scholar] [PubMed]
- Zhang X, Zeng B, Liu Z, Liao Z, Li W, et al. (2014) Comparative diversity of gut microbiota in two different human flora-associated mouse strains. Current Microbiol 69:365-373.
[Crossref] [Google Scholar] [PubMed]
- Zoetendal EG, Raes J, van den Bogert B, Arumagam M, Booijink CC, et al. (2012) The human small intestinal microbiota is driven by rapid intake and conversion of simple carbohydrates. ISME J 6:1415-1426.
[Crossref] [Google Scholar] [PubMed]
- Blaser MJ (2019) Fecal microbiota transplantation for dysbiosis-predictable risks. N Engl J Med 381: 2065-2066. [Crossref]
[Google Scholar] [PubMed]
- Di Bella (2013) Fecal microbiota transplantation: The state of the art. Infect Dis Rep 13: 43-45.
[Crossref] [Google Scholar] [PubMed]
- Khoruts A, Dicksved J, Jansson JK, Sadowsky MJ (2010) Changes in the composition of the human fecal microbiome after bacteriotherapy for recurrent Clostridium difficile-associated diarrhea. J Clin Gastroenterol 44: 354-360.
[Crossref] [Google Scholar] [PubMed]
- Borody TJ, Khoruts A (2012) Fecal microbiota transplantation and emerging applications. Nat Rev Gastroaenterol Hepatol 9: 88-96.
[Crossref] [Google Scholar] [PubMed]
- Cunningham-Rundles S, Ahrné S, Johann-Lang R, Abuav R, Dunn-Navarra A, et al. (2011) Effect of probiotic bacteria on microbial host defense, growth, and immune function in immunodeficiency virus type-1 infection. Nutrients 3: 1042-1070.
[Crossref] [Google Scholar] [PubMed]
- Froebel LK, Jalukar S, Lavergne TA, Lee JT, Duong T (2019) Administration of dietary prebiotics improves growth performance and reduces pathogen colonization in broiler chickens. Poultry Sci 98: 6668-6676.
[Crossref] [Google Scholar] [PubMed]
- Magrone T, Jirillo E(2013) The interaction between gut microbiota and age-related changes in immune function and inflammation. Immun Ageing 10: 31. [Crossref]
[Google Scholar] [PubMed]
- Linninge C, Xu J, Bahl MI, Ahrne S, Molin G (2019) Lactobacillus fermentum and Lactobacillus plantarum increased gut microbiota diversity and functionality, and mitigated Enterobacteriaceae, in a mouse model. Beneficial Microbes 10: 413-424.
[Crossref] [Google Scholar] [PubMed]
- Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, et al. (2010) Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. PNAS 108: 4516-4522.
[Crossref] [Google Scholar] [PubMed]
- Fadrosh DW, Ma B, Gajer P, Sengamalay N, Ott S, et al. (2014) An improved dual-indexing approach for multiplexed 16S rRNA gene sequencing on the Illumina MiSeq platform. Microbiome 1: 6.
[Crossref] [Google Scholar] [PubMed]
- Konstantinidis KT, Tiedje JM (2005) Genomic insights that advance the species definition for prokaryotes. Proc Natl Acad Sci USA 102: 2567-2572.
[Crossref] [Google Scholar] [PubMed]
- Lazarevic V, Whiteson K, Huse S, Hernandez D, Farinelli L, et al. (2009) Metagenomic study of the oral microbiota by Illumina high-throughput sequencing. J Microbiol Meth 79: 266-271.
[Crossref] [Google Scholar] [PubMed]
- Lee JC, Gutell RR (2012) A comparison of the crystal structure of eukaryotic and bacterial SSU ribosomal RNAs reveals common structural features in the hypervariable regions. PLoS ONE 5:1-6.
[Crossref] [Google Scholar] [PubMed]
- Blanton C (2014) Personal communication.
- Huse SM, Welch DM, Morrison HG, Sogin ML (2010) Ironing out the wrinkles in the rare biosphere through improved OUT clustering. Environ Microbiol 12: 1889-1898.
[Crossref] [Google Scholar] [PubMed]
- Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD (2013) Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. App Environ Microbiol 79: 5112-5120.
[Crossref] [Google Scholar] [PubMed]
- Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, et al. (2009) Introducing mother: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol75: 7537-7541.
[Crossref] [Google Scholar] [PubMed]
- Tripathi BM (2014) Spatial effects on soil bacterial communities in malaysian tropical forests. Environ Microbiol.
[Crossref] [Google Scholar] [PubMed]
- Meyer F, Paarmann D, D’Souza M, Olson R, Glass EM, et al. (2008) The metagenomics RAST server- a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics 9:386.
[Crossref] [Google Scholar] [PubMed]
- Edgar RC (2004) Local homology recognition and distance measures in linear time using compressed amino acid alphabets. Nucleic Acids Res. 32: 380-385.
[Crossref] [Google Scholar] [PubMed]
- Schloss PD, Westcott SL (2011) Assessing and improving methods used in operational taxonomic unit-based approaches for 16S rRNA gene sequence analysis. Appl Environ Microbiol 10: 3219-3226.
[Crossref] [Google Scholar] [PubMed]
- Schloss PD (2010) The effects of alignment quality, distance calculation method, sequence filtering, and region on the analysis of 16S rRNA gene-based studies. PLoS Comput Biol 7: e1000844.
[Crossref] [Google Scholar] [PubMed]
- Werner JJ, Zhou D, Caporaso JG, Knight R, Angenent LT (2012) Comparison of Illumina paired-end and single-direction sequencing for microbial 16S rRNA gene amplicon surveys. ISME J 6:1273-1276.
[Crossref] [Google Scholar] [PubMed]
- Schloss PD (2009) A high-throughput DNA sequence aligner for microbial ecological studies. PLoS ONE 12: e8230.
[Crossref] [Google Scholar] [PubMed]
- Bartram AK, Lynch MD, Stearns JC, Moreno-Hagelsieb G, Neufeld JD (2011) Generation of multimillion-sequence 16S rRNA gene libraries from complex microbial communities by assembling paired-end Illumina reads. App Environ Microbiol 77: 3846-3852.
[Crossref] [Google Scholar] [PubMed]
- Schloss PD (2008) Evaluating different approaches that test whether microbial communities have the same structure. ISME J 2: 265-275.
[Crossref] [Google Scholar] [PubMed]
- Liu WT, Marsh TL, Cheng H, Forney LJ (1997) Characterization of microbial diversity by determining terminal restriction fragment length polymorphisms of genes encoding 16S rRNA. Appl Environ Microbiol 63: 4516-4522.
[Crossref] [Google Scholar] [PubMed]
- Blankenberg D, Gordon A, von Kuster G, Coraor N, Taylor J, et al. (2010) Manipulation of FASTQ data with galaxy. Bioinformatics 26: 1783-1785.
[Crossref] [Google Scholar] [PubMed]
- Bӓckhed F, Ding H, Wang T, Hooper LV, Koh GY, et al. (2004) The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci USA 101: 15718-15723.
[Crossref] [Google Scholar] [PubMed]
- Chen J, He X, Huang J (2014) Diet effects in gut microbiome and obesity. J Food Sci 79:R442-R451.
[Crossref] [Google Scholar] [PubMed]
- Clement BG, Kehl LE, Debord KL, Kitts CL (1998) Terminal restriction fragment patterns (TRFPs), a rapid, PCR-based method for the comparison of complex bacterial communities. J Microbiol Met 31:135-142.
[Google Scholar] [PubMed]
- Cock PJ, Fields CJ, Goto N, Heuer ML, Rice PM (2009) The sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Res 6:1767-1771.
[Crossref] [Google Scholar] [PubMed]
- Costello EK, Lauber CL, Hamady M, Fierer N, Gordon JI, et al. (2009) Bacterial community variation in human body habitats across space and time. Science 326: 1694-1697.
[Crossref] [Google Scholar] [PubMed]
- de Wit N, Derrien M, Bosch-Vermeulen H, Oosterink E, Keshtkar S, et al. (2012) Saturated fat stimulates obesity and hepatic steatosis and affects gut microbiota composition by an enhanced overflow of dietary fat to the distal intestine. Am J Physiol Gastrointest Liver Physiol 303: G589-599.
[Crossref] [Google Scholar] [PubMed]
- Imaizumi K, Akutsu T, Miyasaka S, YoshinoM (2007) Development of species identification tests targeting the 16S ribosomal RNA coding region in mitochondrial DNA. International J Legal Med 121:184-191
[Crossref] [Google Scholar] [PubMed]
- Kostic AD, Howitt MR, Garrett WS (2013) Exploring host-microbiota interactions in animal models and humans. Genes Dev 27: 701-718.
[Crossref] [Google Scholar] [PubMed]
- Ley RE, Backhed F, Turnbaugh P, Lozupone CA, Knight RD, et al. (2005) Obesity alters gut microbial ecology. Proc Natl Acad Sci USA 102: 11070-11075.
[Crossref] [Google Scholar] [PubMed]
- McGarr SE, Ridlon JM, Hylemon PB (2005) Diet, anaerobic bacterial metabolism, and colon cancer: a review of the literature. J Clin Gastroenterol 39: 98-109.
[Google Scholar] [PubMed]
- Nicholson JK, Holmes E, Wilson ID (2005) Gut microorganisms, mammalian metabolism and personalized health care. Nat Rev Microbiol 3: 431-438.
[Crossref] [Google Scholar] [PubMed]
- Noverr MC, Huffnagle GB (2005) The ‘microflora hypothesis’ of allergic responses. Clin Exp Allergy 35:1511-1520.
[Crossref] [Google Scholar] [PubMed]
- Reddy BVB, Kallifidas D, Kim JH, Harlop-Powers Z, Feng Z, et al. (2012) Natural product biosynthetic gene diversity in geographically distinct soil microbiomes. Appl Environ Microbiol 78: 3744-3752.
[Crossref] [Google Scholar] [PubMed]
Citation: Christensen LM, Blanton C, Sheridan PP (2024) Paired-End Phylogenetic Analysis of Bacterial Population Differences in Mus Musculus Cecum in Response to Synbiotic Administration. J Infect Dis Ther 12: 577. DOI: 10.4172/2332-0877.1000577
Copyright: © 2024 Christensen LM, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Select your language of interest to view the total content in your interested language
Share This Article
Recommended Journals
Open Access Journals
Article Tools
Article Usage
- Total views: 1978
- [From(publication date): 0-2024 - Dec 07, 2025]
- Breakdown by view type
- HTML page views: 1633
- PDF downloads: 345