K16ApoE Enhances A β-associated 11C-PiB Deposition and PET Signal in APP/ PS1 Transgenic Mice
Received: 29-May-2019 / Accepted Date: 13-Jun-2019 / Published Date: 20-Jun-2019 DOI: 10.4172/2161-0460.1000468
Abstract
Objective: Transgenic mouse models are central to the study of Alzheimer’s disease and aid in elucidating the underlying pathophysiology. Mouse models also provide a system in which to test potential therapeutic strategies. PET imaging plays a central clinical role in diagnosing human cases of Alzheimer’s disease but has had variable performance in mouse models. We investigated the potential role of the K16ApoE carrier peptide to enhance delivery of a radiolabeled PET imaging tracer, 11C-PiB and assess whether this corresponds to improved sensitivity of the PET modality in APP/PS1 transgenic mice.
Methods: Brain-delivery of 11C-PiB was accomplished by sequential bolus injections of K16ApoE and 11C-PiB via femoral vein injections. Distribution of 11C-PiB to the brain and heart was quantified via dynamic PET/CT imaging and digital autoradiography.
Results: K16ApoE carrier peptide increased the brain uptake of 11C-PiB in both wild-type and APP/PS1 mice. Administration of K16ApoE increased the PET standard uptake value of 11C-PiB at 5 minutes in WT mice from 1.132 to 2.963 (p=0.006) and in APP/PS1 mice from 0.842 to 3.268 (p=0.016). Enhancement peaked at 5 minutes. Binding was reversible as demonstrable by Logan plots with similarly increased kinetics in both WT and APP/PS1 mice. The absolute values were higher in APP/PS1 mice suggesting increased retention. The increased retention in APP/PS1 mice was consistent with specific binding to Aβ plaques as unlabeled PiB showed competitive reduction of 11C-PiB signal retention.
Conclusion: K16ApoE-mediates enhancement of 11C-PiB signal in APP/PS1 mice brains with increase in the PET sensitivity. There is increased uptake kinetics in both WT and APP/PS1 mice with specific retention due to Aβ plaque binding in the latter. This improved sensitivity of PET scanning in the APP/PS1 transgenic mouse model. Such enhanced delivery of this PET tracer has implications for development and testing of new hypotheses and the efficacy of novel therapeutic paradigms.
Keywords: Alzheimer’s disease; 11C-PiB, PET; Blood-brain barrier; Carrier peptide; Digital autoradiography; DVR; RRT; Mouse model
Introduction
Alzheimer’s disease (AD) is a neurodegenerative disease histopathologically characterized by the presence of neuritic plaques and neurofibrillary tangles in the brain of afflicted patients. Pathophysiologically, these changes are associated with the deposition of amyloid-β (Aβ), hyperphosphorylation of tau proteins, and neuroinflammation and glial activation. The neurocognitive decline parallels the degree of histopathological changes [1]. Given the hypothesized role in the pathophysiology of AD and the correlation with disease progression, Aβ plaques are important targets for the early diagnosis and treatment of AD [2-4]. Definitive diagnosis of AD requires autopsy but the recent development of Positron Emission Tomography (PET) radiotracers with affinity for Aβ allow for detection of several key pathological indicators of AD in living human subjects [5,6].
Neuroimaging has enabled in vivo visualization of pathological changes in the brain associated with AD and has improved our understanding of the natural history of these changes. The 11C-labeled Pittsburg compound B (11C-PiB), a benzothiazole derivative analog of Thioflavin T, is a widely used PET radiotracer in AD [7,8]. It binds Aβ plaques with high affinity but ultimately, sensitivity is dependent on the plaque burden and degree of radiotracer uptake [7,9-11]. The degree of radiotracer uptake is a manipulable variable and both the enhanced delivery of radiotracer to the brain and the extent of plaque binding are amenable to methodological intervention. Enhanced brain delivery of a radiotracer, such as 11C-PiB, could enable the detection of plaque-load in AD brain with greater sensitivity with potential implications for earlier diagnosis of AD. As AD has a long prodromal phase, earlier diagnosis may lead to more effective use of disease-modifying agents.
Mouse models play a central role in elucidating the pathophysiology of AD and have been heavily used to evaluate candidate therapeutic strategies. The latter requires longitudinal evaluation of individual mice pre- and post-treatment. Small animal PET scanners facilitate noninvasive longitudinal imaging when used with appropriately selected PET tracers and is a potentially powerful tool for discovery and therapy monitoring [12]. However, uptake of radiotracers have been inconsistent and often inadequate [13-15]. The sensitivity of detection 11C-PiB in particular, is a function of the specific mouse model which in turn affects the underlying structure of the Aβ plaque [16].
We synthesized a novel carrier peptide, K16ApoE (a polymer of 16 lysine residues bound to the low-density lipoprotein receptor-binding sequence of apolipoprotein E), and demonstrated its ability to transport both large molecules (such as enzymes and immunoglobulins) and small molecules (such as Evans Blue and cisplatin) across the Blood- Brain Barrier (BBB) in the absence of a covalent linkage [17-20]. We hypothesized that K16ApoE could similarly enhance delivery of 11C-PiB across the BBB, leading to enhanced Aβ plaque-associated signal in an APP/PS1 transgenic mouse model.
Materials and Methods
Animal strains and use
11C-PiB PET imaging was conducted in BALB/c Wild-Type (WT) and APP/PS1 (AD) female mice. The double-transgenic AD mice were cross-bred in-house. Hemizygous transgenic mice (mouse strain: C57B6/SJL; ID no. Tg2576) expressing mutant APP 695 containing a double mutation (K670N,M671L) [21] were mated with homozygous transgenic mice (mouse strain: Swiss Webster/B6D2; ID no.M146L6.2) expressing mutant human PS1 with an M146L point mutation [22]. All mice were housed in a dedicated mouse facility. Animals were under the care of a licensed veterinarian and were inspected on a daily basis. There was ad libitum access to soy-free chow and water. The study was approved by the Mayo Clinic Institutional Animal Care and Use Committee (IACUC) and all applicable institutional and national guidelines for the care and use of laboratory animals were followed.
Synthesis of 11C-PiB and 11C-Choline
11C-PiB and 11C-Choline were synthesized on site at the Mayo Clinic Cyclotron Facility as previously described [17,19]. Average specific activity of 11C-PiB was 7.72 ± 6.42 Ci/μmol. 11C-Choline was synthesized by labeling dimethylethanolamine with 11C-methyliodine and formulated for injection in saline.
K16ApoE-mediated administration of 11C-PiB and PET imaging
K16ApoE was injected into the femoral vein followed by the injection of 11C-PiB (621+/-45μCi) after a 10 minute interval as our group previously described [20]. Given the short half-life (20 min) of 11C-PiB, the tracer was also injected immediately following injection of the polypeptide carrier in order to capture the time-period when the radioactive signal may be at its peak. 11C-PiB-PET scans were performed with an Inveon multimodality PET/CT scanner (Siemens Medical Solutions, USA, Knoxville, TN) with animals under isofluorane anesthesia for the duration of scanning. Initial CT images were acquired at 180 projections with 300 μsec per projection (bin 4, 60 keV, 500μA). Animals were then transitioned to the PET scanner and injections were performed within 90 seconds after initiation of the scan which was acquired over 40 min.
A subset of mice was imaged twice: once after administering 11C-PiB alone and then following co-injection of K16ApoE and 11C-PiB. The first scan was completed as stated above by injecting 11C-PiB alone. After 120 min (~5 half-lives of 11C) following the initial 11C-PiB injection and scanning, a combination of 11C-PIB and K16ApoE was injected and the second scan was repeated with the same parameters. Upon completion of scanning, the mice were deeply anesthetized then sacrificed via cardiac puncture.
K16ApoE-mediated administration of 11C- Choline and PET imaging
PET imaging with 11C-choline was performed to assess perfusionrelated increase in brain uptake that could potentially confound increased binding of 11C-PiB. The 11C-choline show very little binding to the normal brain tissue and mostly exhibit perfusion limited brain delivery. Hence, 11C-choline will help verify if K16ApoE enhances the brain uptake of small molecular tracers by increasing the brain perfusion. Mice were injected with 300 μg of K16ApoE followed by 11C-Choline (571.63+23.99 μCi in 200 μL).
Analysis of PET imaging data
The 40-minute PET acquisition list mode data was reconstructed into 5-minute frames. The first 10 minutes of the acquisition was also reconstructed into 1-minute frames. The PET/CT data was analyzed using PMOD Biomedical Image Quantification and Kinetic Modeling Software (PMOD Technologies, Switzerland). A volume of interest was drawn over the brain using the CT scan to delineate the bony anatomy. A uniform size background region was drawn in the left shoulder muscle. The activity concentration of the background region was subtracted from the activity concentration of the brain to correct for the background. Standard Uptake Values (SUV) was normalized to body weight.
Determination of distribution volume ratio (DVR) and relative residence time (RRT) of 11C-PiB delivered to the brain
To assess the kinetics of 11C-PiB uptake in the brain with and without K16ApoE, DVR and RRT were calculated from the PET data. PET data was first quantified by multiple-time graphical analysis. A Time-Activity Curve (TAC) of the injected radiolabeled tracer revealed that much of the tracer is rapidly washed out of the brain (data not shown), consequently, a Logan plot was generated to determine the kinetics of brain uptake of 11C-PiB [23]. The Logan plot was created by plotting:
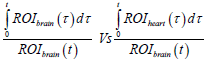
Where, ROIbrain describes the radioactivity of 11C-PiB captured by PET imager in the brain at time τ. The ROIheart is the radioactivity of 11C-PiB in the reference tissue (heart) at time τ, whereas t is the time elapsed between the tracer administration and a given time-frame of analysis.
A linear plot was obtained, which is described by the following equation:
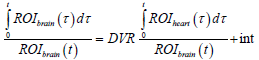
The slope of this plot indicates DVR [23]. It is the ratio of distribution volume of the tracer in the brain to the distribution volume of the tracer in the reference tissue (heart). The intercept (int) of the plot indicates the RRT of 11C-PiB in brain compared with the chosen reference tissue (heart).
Digital autoradiography and quantification of Aβ deposits from digital autoradiograms
Brains were flash frozen in dry ice-chilled isopentane and sectioned. Slices were imaged on a BAS-TR2025 machine and scanned with a BAS-5000 scanner. Ten brain sections per mouse were analyzed via digital autoradiography via creation of digital autoradiograms of all mice brains with signal measured in digital light units (DLU). DLU/ mm2 values (Table 1, Column 2) represent an average of 180 DLU values obtained from 3 mice brains that received 11C-PiB only (3 brains × 10 slices/brain × 6 spots/slice), and an average of 360 DLU values from brains in mice given 11C-PiB with K16ApoE (6 brains × 10 slices/ brain × 6 spots/slice). Fold change (Table 1, Column 3) was obtained by dividing the mean DLU/mm2 value obtained with K16ApoE by the mean DLU/mm2 value obtained 11C-PiB only.
| Group | DLU/mm2 (mean ± SD) | Fold change (p value) | Global DLU (mean ± SD) | Fold change (p value) |
|---|---|---|---|---|
| Without K16ApoE | 670.14 ± 128.24 | 14.12 (1.21E-13) | 30202.78 ± 5110.24 | 12.01 (9.14E-14) |
| With K16ApoE | 9460.37 ± 1417.91 | 326300.47 ± 42151.25 |
Mean global DLU values (Column 4) represent an average of 30 DLU values obtained from 3 mice brains receiving 11C-PiB without K16ApoE (3 brains x 10 slices/brain x one whole brain area/slice), and 60 DLU values corresponding to six mice brains receiving 11C-PiB with K16ApoE (6 brains x 10 slices/brain x one whole brain area/slice). Fold change (Column 5) was obtained by dividing the mean 11C-PiB global DLU value obtained with K16ApoE by the mean 11C-PiB global DLU value obtained without K16ApoE.
Table 1: Quantification of brain uptake of 11C-PiB in AD mice with and without carrier (K16ApoE) using DLU values obtained from digital autoradiograms
K16ApoE-mediated 11C-PiB competitive assay
K16ApoE was injected as per above followed by injection of unlabeled PiB. After a 5-minute delay, 11C-PiB (621+/-45Ci) was administered and the animal underwent PET imaging as per above. As a control, K16ApoE was injected followed by injection of 11C-PiB (621+/-45μCi).
Immunohistochemical and thioflavin S staining of amyloid plaques
Immunohistochemical staining of the Aβ plaques in the mouse brain was performed with thioflavin S and 4G8 anti-APP mouse monoclonal antibody (Covance Research Products, Berkeley, CA) as previously described [12,20].
Data and statistics
Results are presented as mean ± standard deviation unless otherwise stated. Absolute SUVs were compared using the two-tailed student’s t-test.
Results
K16ApoE enhances brain delivery of 11C-PiB in both WT and APP/PS1 mice
Administration of K16ApoE increased the SUV of 11C-PiB at 5 minutes in WT mice from 1.132 to 2.963 (p=0.006) and in AD mice from 0.842 to 3.268 (p=0.016) as depicted in Figure 1. Brain uptake peaked at 5 min with subsequent decline over the 40-minute monitoring period. Injection of 11C-PiB immediately following K16ApoE administration produced greater brain-uptake compared to that seen after a 10 min delay. As summarized in Table 2, K16ApoE enhanced brain uptake of 11C-PiB in both WT and AD mice but the enhancement was more pronounced in AD compared to WT mice.
| Time | WT (Average SUV) | AD (Average SUV) | ||||
|---|---|---|---|---|---|---|
| (min) | 11C-PiB Only | 11C-PiB 0 min after K16ApoE | 11C-PiB 10 min after K16ApoE | 11C-PiB Only | 11C-PiB 0 min after K16ApoE | 11C-PiB 10 min after K16ApoE |
| 5 | 1.13 | 2.8 | 1.39 | 1.09 | 3.26 | 1.23 |
| 10 | 0.42 | 1.79 | 1.43 | 0.63 | 2.16 | 1.33 |
| 15 | 0.17 | 0.87 | 0.88 | 0.34 | 1.28 | 0.88 |
| 20 | 0.09 | 0.51 | 0.58 | 0.21 | 0.91 | 0.64 |
| 25 | 0.04 | 0.33 | 0.4 | 0.15 | 0.7 | 0.5 |
| 30 | 0.03 | 0.22 | 0.28 | 0.11 | 0.57 | 0.43 |
| 35 | 0.02 | 0.18 | 0.23 | 0.09 | 0.48 | 0.38 |
| 40 | 0.01 | 0.15 | 0.18 | 0.08 | 0.42 | 0.34 |
Table 2: Time course of brain standard uptake values of 11C-PiB in WT and AD mice with and without pre-infusion of the K16ApoE peptide carrier

Figure 1: K16ApoE enhances delivery of 11C-PiB in WT (A) and AD (B) mice brains by PET imaging. 11C-PiB was delivered without the carrier peptide, K16ApoE (blue bars); 10 min after injection of K16ApoE (red bars); and 0 min after injection of K16ApoE (green bars). Insets show two representative images of mice heads without (top) and with (bottom) K16ApoE (no delay). Six mice were evaluated in each group. Bars represent mean standard uptake values and error bars are standard deviation.
These investigations were also conducted employing a crossover study design using 3 WT and 4 AD mice, where each animal was used as ‘control’ (11C-PiB alone) and ‘test’ (11C-PiB plus K16ApoE) after a washout period of 120 minutes. K16ApoE enhanced brain delivery of 11C-PiB ~2.5-fold compared to 11C-PiB alone (Figure 2). Levels of 11C-PiB were not significantly different between the groups after 15 minutes suggesting that K16ApoE may increase the kinetics of early uptake but not the retention of brain 11C-PiB levels.

Figure 2: K16ApoE-mediated enhanced delivery of 11C-PiB remains robust by PET imaging in crossover design. WT (A) and AD (B) mice received 11C-PiB alone (hashed bars). After a 120-minute washout period, mice received K16ApoE and 11C-PiB without delay between administrations (solid bars). Insets show two representative images each of WT and AD mice heads without (top) and with (bottom) K16ApoE. Three wildtype (P28, P29 and P30) and four AD (P31, P32, P37 and P38) mice were evaluated. Bars represent mean standard uptake values and error bars are standard deviation.
11C-PiB brain-uptake kinetics in WT and AD mice
Time-Activity Curves (TAC) generated from multiple-time graphical analyses of the 11C-PiB indicated that the tracer (unbound and/or reversibly bound) is rapidly washed out from the brain (data not shown). Thus, Logan plots of the tracer in AD (Figure 3A) and WT (Figure 3D) mice were generated to quantify the Distribution Volume Ratio (DVR) and Relative Residence Time (RRT) of the tracer. The slopes of the Logan plots represent the DVR for AD (Figure 3B) and WT mice (Figure 3E), while the intercepts indicate the RRT for AD (Figure 3C) and WT (Figure 3F) mice. There was a significant increase in the DVR of 11C-PiB administered immediately after the K16ApoE carrier peptide. Mean DVR was 1.116±0.056 and 0.823±0.075 for AD and WT mice, respectively. In the presence of K16ApoE, DVR increased to 2.386±0.277 and 1.92±0.231 for AD and WT mice, respectively and represents a respective increase of 113.8% and 133.4%. DVR did not increase in either WT or AD mice when 11C-PiB administration was delayed by 10 min after K16ApoE was given (data not shown).

Figure 3: K16ApoE enhances 11C-PiB uptake kinetics in WT and AD mice. (A, D) Logan plots representing brain uptake of 11C-PiB with (closed squares/circles) and without (open squares/circles) K16ApoE in AD mice (A) and WT (D) mice. Distribution volume ratio (DVR) of 11C-PiB in AD (B) and WT (E) are increased in the presence of K16ApoE. Similarly, the relative residence time (RRT) for 11C-PiB in AD (C) and WT (F) mice are increased. *represent p-values < 0.001 as calculated by a two-tailed student’s t-test.
There was also a significant increase in the RRT of 11C-PiB in WT and AD mice when 11C-PiB was administered immediately after injecting the carrier peptide, K16ApoE. In AD mice, the RRT increased from 0.748 ± 0.217 min to 3.088 ± 0.559 min (312.8%) when the carrier peptide was given prior to 11C-PiB and 0.269 ± 0.069 min to 2.528 ± 0.356 min (839.8%) in WT animals.
Digital quantification of plaque-associated 11C-PiB in the brain
Digital autoradiograms of brain slices from WT and AD brains confirm enhanced delivery of 11C-PiB in the presence of K16ApoE with a more robust signal increase in AD versus WT mice (Figure 4). Quantification of the digital autoradiogram signal was performed on an entire slice (global DLU) or from six visually-intense areas to generate DLU/mm2 (Supplementary Figure 1). Global DLU and DLU/ mm2 showed greater increase in signal intensity in the AD brain when 11C-PiB was delivered with K16ApoE. A comprehensive analysis of the DLU/mm2 data show >14-fold (p=1.211E-13) and analysis of ‘global DLU’ data show >12-fold (p=9.137E-14) greater retention of 11C-PiB in AD mice brains when administered with K16ApoE (Table 1). Aβ plaques were found in the AD mouse brains as ascertained by Thioflavin S staining and by immunohistochemistry and correlated with areas of increased K16ApoE-mediated increased 11C-PiB signal (Figure 5).

Figure 4: K16ApoE increases 11C-PiB signal by digital autoradiography ex vivo. Panel shows three representative digital chromatograms of mice brain slices per group. There is increased signal in AD mice following K16ApoE coadministration.

Figure 5: K16ApoE-mediated increase in 11C-PiB uptake overlaps with Aß plaques in AD mice. Aß plaques in WT (left panel) and AD (right panel) mice were assessed by autoradiography for 11C-PiB in the absence (A, B) and presence (C, D) of K16ApoE carrier peptide. Global and regional DLU values are shown beneath each panel for objective comparison. There is dramatic increase in DLU signal in AD mice when K16ApoE is co-administered. Thioflavin S staining (E, F) and 4G8 anti-APP mouse monoclonal antibody immunohistochemistry (G, H) in WT (E, G) and AD (F, H) mice in the absence of K16ApoE carrier peptide show increased Aß-plaques in AD versus WT mice and corresponds to areas of 11C-PiB autoradiography signal.
Determination of specificity of PiB delivery and plaque binding in AD mice
To determine whether the enhanced binding of 11C-PiB facilitated by K16ApoE was due to site-specific interactions, unlabeled PiB was administered to AD mice in the presence of K16ApoE followed by the administration of 11C-PiB and the signal uptake was examined by PET PET-imaging as described above. There was a ~3-fold decrease in SUV when 11C-PiB was injected following unlabeled PiB (Figure 6A) suggesting that the interaction was saturable. This reduction was not seen in WT mice (data not shown).

Figure 6: K16ApoE-mediated brain uptake of 11C-PiB is a specific process likely dependent on Aß plaques binding. (A) Unlabeled PiB reduces K16ApoE-mediated brain uptake of 11C-PiB in AD mice. Brain-uptake of 11C-PiB was evaluated via PET-mediated quantification in the presence (blue bars) or absence (red bars) of unlabeled PiB. Inset shows representative brain slices in the presence (top) or absence (bottom) of unlabeled PiB. (B) K6ApoE does not increase 11C-Choline levels by autoradiography. K16ApoE-mediated delivery of 11C-Choline in WT (top) and AD (middle) mice are shown. For comparison, K16ApoE significantly enhances uptake and retention of 11C-PiB.
To determine if the presence of amyloid plaques is the reason underlying the increased retention of 11C-PiB in AD mouse brains, which is evident from the increased signal on autoradiograms, we reasoned that an agent such as choline, which is not known to bind to amyloid plaques, would maintain similar signal intensities in AD and WT mouse brains. To evaluate this premise, we delivered 11C-Choline to WT as well as AD mice with and without K16ApoE. Autoradiography confirmed similar 11C-Choline distribution in WT and AD mouse brains (Figure 6B).
Discussion
11C-PiB has been remarkably effective in assessing amyloid plaque burden in human brains but its ability to detect amyloid plaques in AD transgenic mouse models remains tenuous [9,16]. The inconsistent performance of 11C-PiB in detecting amyloid deposits in transgenic mouse models has been attributed to low specific binding of the tracer to amyloid plaques generated in mouse models secondary to a paucity of high-affinity binding sites [9,24] as well as presumed differences in amyloid plaque morphology across transgenic models [25,26]. While these factors alter the extent of 11C-PiB binding to amyloid plaques and engender variations in PET signal, the contribution of brain uptake of 11C-PiB across the BBB in humans versus transgenic mice is also a significant factor; physiological differences between humans and mice lead to variations in the brain uptake kinetics of various compounds including 11C-PiB. These kinetic processes could be differentially impacted in AD and may exacerbate differences in PiB distribution in APP/PS1 transgenic mice [27-29]. Therefore, we hypothesized that the variability in 11C-PiB signal reported in AD transgenic mouse brain could also be improved by enhanced BBB permeability by K16ApoE.
K16ApoE is a novel carrier peptide that was shown to transiently increase the permeability of a diverse array of large and small molecules to the brain [20,30-33]. Our studies demonstrate a robust K16ApoEmediated increase in 11C-PiB in both WT and AD mice. As anticipated, the signal was greater in the APP/PS1 transgenic mice compared to WT mice as anticipated given a higher burden of Aβ in the former. We also noted that the brain uptake of 11C-PiB is greater when it is delivered immediately following the administration of K16ApoE, rather than administering following a 10 min delay. This observation suggests that K16ApoE may act transiently to increase the BBB permeability, and enhance CNS delivery of 11C-PiB.
Brain-uptake kinetics of PET imaging ligands is routinely quantified by multiple-time graphical analysis. In this study, the PET data obtained from animals administered with 11C-PiB alone or in conjunction with K16ApoE was best fitted by Logan graphical analysis indicating that the tracer binding is reversible. The DVR parameter measures changes in binding to a physiological target, while the RRT parameter measures the residence time of 11C-PiB in brain compared to the heart vis-à-vis, the reference tissue. As K16ApoE administration increased both the DVR and RRT of 11C-PiB in both WT and AD mice, we infer that the carrier peptide increased binding of 11C-PiB to the brain tissue in both WT and AD mice. The binding of 11C-PiB in WT mouse brain is not surprising due to the reported propensity of 11C-PiB to bind nonspecifically to the white matter nonspecifically [34]. Since K16ApoE-mediated relative increases of DVR and RRT of 11C-PiB in both WT and AD mice are similar but with larger absolute 11C-PiB DVR and RRT values in AD versus WT mice, K16ApoE likely enables greater retention of 11C-PiB in AD brains in a process dependent on Aβ specific binding. This occurs without impact on delivery kinetics in WT and AD mice, per se.
Specificity of the binding in AD mice was investigated by prebinding of unlabeled PiB. This reduced the uptake of 11C-PiB in AD but not WT mice suggesting that the increased uptake can be reduced by competitive binding and suggests specific, saturable interactions of 11C-PiB with Aβ. When 11C-choline, a compound with no known binding to Aβ was administered in the presence of carrier, there was no appreciable increase in uptake or retention in AD compared to WT mice. Taken together, these results suggest that the K16ApoE-mediated increased uptake of 11C-PiB in AD compared to WT mice is a specific process and likely mediated by Aβ.
It is notable that when quantified via digital autoradiograms, 11C-PiB show >10-fold brain-retention of 11C-PiB in AD mice brains when delivered with K16ApoE than without. The increase in 11C-PiB retention assessed by autoradiography is much more than that observed with PET-imaging (~3-fold). This difference is probably due to the inability of PET imaging to differentiate 11C-PiB specifically bound to the plaques from that bound nonspecifically to white matter. The processing steps employed in autoradiography, could remove most of the nonspecifically bound label in the brain, so that the label specifically bound to amyloid plaques could be captured. Thus, autoradiography indicates that K16ApoE increases the 11C-PiB signal in AD brains, most likely by enhancing the amount of 11C-PiB specifically bound to amyloid plaques in the brain. This could be substantiated by the observation that unlabeled PiB was able to reduce the binding of 11C-PiB to the plaques, and that an irrelevant PET probe 11C-Choline that does not bind to amyloid plaques, did not shown any enhancement in the signal.
Conclusion
In summary, we show that a 36-amino acid synthetic peptide, previously shown to enhance the transport of various molecules to the brain, can greatly enhance the brain uptake of 11C-PiB in AD mice brains facilitating amyloid plaque detection. These results could have important implication for earlier detection of amyloid plaque burden in the transgenic mouse models by PET which is a current limitation of the utility of AD mouse models [12]. This may assist with monitoring the efficacy of novel therapeutic compounds and significantly expedite AD drug discovery and development. This strategy may also enhance brain delivery of therapeutic agents such as anti-amyloid antibodies that have shown some efficacy in the clinical trials. Indeed, our group has demonstrated efficacy of the K16ApoE peptide in nanoparticle targeting of Aβ plaques [31] as well as enhanced delivery of a number of chemotherapeutic agents [20]. This suggests that the role of K16ApoE in enhancing delivery of substances across the BBB has far-reaching implications for a wide array of neurologic diseases.
Acknowledgement
This work was funded by the Mayo Clinic. We thank Ping Fang for synthesizing 11C-PiB.
Conflict of Interest
Dr. V. Lowe serves as a consultant for Bayer Schering Pharma, Philips Molecular Imaging, Piramal Imaging and GE Healthcare and receives research support from GE Healthcare, Siemens Molecular Imaging, AVID Radiopharmaceuticals, the NIH (NIA, NCI), and the MN Partnership for Biotechnology and Medical Genomics. Other authors declare no conflict of interest. The K16ApoE carrier peptide is the subject of an ongoing patent application by the Mayo Clinic.
References
- Lee JC, Kim SJ, Hong S, Kim Y (2019) Diagnosis of Alzheimer’s disease utilizing amyloid and tau as fluid biomarkers. Exp Mol Med 51: 53.
- Ballard C, Gauthier S, Corbett A, Brayne C, Aarsland D, et al. (2011) Alzheimer’s disease. Lancet 377: 1019-1031.
- Chinthapalli K (2014) Alzheimer’s disease: Still a perplexing problem. BMJ 349: g4433.
- Saidlitz P, Voisin T, Vellas B, Payoux P, Gabelle A, et al. (2014) Amyloid imaging in Alzheimer’s disease: A literature review. J Nutr Health Aging 18: 723-740.
- Rinne JO, Brooks DJ, Rossor MN, Fox NC, Bullock R, et al. (2010) 11C-PiB PET assessment of change in fibrillar amyloid-beta load in patients with Alzheimer’s disease treated with bapineuzumab: a phase 2, double-blind, placebo-controlled, ascending-dose study. Lancet Neurol 9: 363-372.
- Nordberg A, Rinne JO, Kadir A, Långström B. (2010) The use of PET in Alzheimer disease. Nat Rev Neurol 6: 78-87.
- Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, et al. (2004) Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol 55: 306-319.
- Rowe CC, Ackerman U, Browne W, Mulligan R, Pike KL, et al. (2008) Imaging of amyloid beta in Alzheimer’s disease with 18F-BAY94-9172, a novel PET tracer: Proof of mechanism. Lancet Neurol 7: 129-135
- Klunk WE, Lopresti BJ, Ikonomovic MD, Lefterov IM, Koldamova RP, et al. (2005) Binding of the positron emission tomography tracer Pittsburgh compound-B reflects the amount of amyloid-beta in Alzheimer’s disease brain but not in transgenic mouse brain. J Neurosci 25: 10598-606.
- Klunk WE, Wang Y, Huang G, Debnath ML, Holt DP, et al. (2003) The binding of 2-(4’-methylaminophenyl)benzothiazole to postmortem brain homogenates is dominated by the amyloid component. J Neurosci 23: 2086-92.
- Mathis CA, Wang Y, Holt DP, Huang G-F, Debnath ML, et al. (2003) Synthesis and evaluation of 11C-labeled 6-substituted 2-arylbenzothiazoles as amyloid imaging agents. J Med Chem 46: 2740-2754.
- Bouter C, Bouter Y (2019) 18F-FDG-PET in Mouse Models of Alzheimer’s Disease. Front Med (Lausanne) 6: 71.
- Cavanaugh SE, Pippin JJ, Barnard ND (2014) Animal models of Alzheimer disease: Historical pitfalls and a path forward. ALTEX 31: 279-302.
- Wengenack TM, Whelan S, Curran GL, Duff KE, Poduslo JF (2000) Quantitative histological analysis of amyloid deposition in Alzheimer’s double transgenic mouse brain. Neuroscience 101: 939-944.
- Waldron A-M, Wyffels L, Verhaeghe J, Bottelbergs A, Richardson J, et al. (2015) Quantitative μPET Imaging of Cerebral Glucose Metabolism and Amyloidosis in the TASTPM Double Transgenic Mouse Model of Alzheimer’s Disease. Curr Alzheimer Res 12: 694-703.
- Snellman A, López-Picón FR, Rokka J, Salmona M, Forloni G, et al. (2013) Longitudinal amyloid imaging in mouse brain with 11C-PIB: comparison of APP23, Tg2576, and APPswe-PS1dE9 mouse models of Alzheimer disease. J Nucl Med 54: 1434-1441.
- Meng Y, Sohar I, Sleat DE, Richardson JR, Reuhl KR, et al. (2014) Effective intravenous therapy for neurodegenerative disease with a therapeutic enzyme and a peptide that mediates delivery to the brain. Mol Ther 22: 547-553.
- Sands MS (2014) A Hitchhiker’s guide to the blood-brain barrier: In trans delivery of a therapeutic enzyme. Mol Ther 22: 483-484.
- Sarkar G, Curran GL, Mahlum E, Decklever T, Wengenack TM, et al. (2011) A carrier for non-covalent delivery of functional beta-galactosidase and antibodies against amyloid plaques and IgM to the brain. PLoS ONE 6: e28881.
- Sarkar G, Curran GL, Sarkaria JN, Lowe VJ, Jenkins RB (2014) Peptide carrier-mediated non-covalent delivery of unmodified cisplatin, methotrexate and other agents via intravenous route to the brain. PLoS ONE 9: e97655.
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, et al. (1996) Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 274: 99-102.
- Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S, et al. (1998) Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med 4: 97-100.
- Logan J, Fowler JS, Volkow ND, Wolf AP, Dewey SL, et al. (1990) Graphical analysis of reversible radioligand binding from time-activity measurements applied to [N-11C-methyl]-(-)-cocaine PET studies in human subjects. J Cereb Blood Flow Metab 10: 740-747.
- Toyama H, Ye D, Ichise M, Liow J-S, Cai L, et al. (2005) PET imaging of brain with the beta-amyloid probe, [11C]6-OH-BTA-1, in a transgenic mouse model of Alzheimer’s disease. Eur J Nucl Med Mol Imaging 32: 593-600.
- Maeda J, Ji B, Irie T, Tomiyama T, Maruyama M, et al. (2007) Longitudinal, quantitative assessment of amyloid, neuroinflammation, and anti-amyloid treatment in a living mouse model of Alzheimer’s disease enabled by positron emission tomography. J Neurosci 27: 10957-10968.
- Ye L, Morgenstern JL, Lamb JR, Lockhart A (2006) Characterisation of the binding of amyloid imaging tracers to rodent Abeta fibrils and rodent-human Abeta co-polymers. Biochem Biophys Res Commun 347: 669-77.
- Caserta MT, Caccioppo D, Lapin GD, Ragin A, Groothuis DR (1998) Blood-brain barrier integrity in Alzheimer’s disease patients and elderly control subjects. J Neuropsychiatry Clin Neurosci 10: 78-84.
- Starr JM, Farrall AJ, Armitage P, McGurn B, Wardlaw J (2009) Blood-brain barrier permeability in Alzheimer’s disease: A case-control MRI study. Psychiatry Res 171: 232-41.
- Poduslo JF, Curran GL, Wengenack TM, Malester B, Duff K (2001) Permeability of proteins at the blood-brain barrier in the normal adult mouse and double transgenic mouse model of Alzheimer’s disease. Neurobiol Dis 8: 555-67.
- Lu L, Michelena TM, Wong A, Zhang CJ, Meng Y (2018) The inhibition of acetylcholinesterase by a brain-targeting polylysine-ApoE peptide: Biochemical and structural characterizations. Conf Proc IEEE Eng Med Biol Soc 2018: 155-158.
- Ahlschwede KM, Curran GL, Rosenberg JT, Grant SC, Sarkar G, et al. (2019) Cationic carrier peptide enhances cerebrovascular targeting of nanoparticles in Alzheimer’s disease brain. Nanomedicine 16: 258-66.
- Zou Z, Shen Q, Pang Y, Li X, Chen Y, et al. (2019) The synthesized transporter K16APoE enabled the therapeutic HAYED peptide to cross the blood-brain barrier and remove excess iron and radicals in the brain, thus easing Alzheimer’s disease. Drug Deliv Transl Res 9: 394-403.
- Meng Y, Wiseman JA, Nemtsova Y, Moore DF, Guevarra J, et al. (2017) A Basic ApoE-Based Peptide Mediator to Deliver Proteins across the Blood-Brain Barrier: Long-Term Efficacy, Toxicity, and Mechanism. Mol Ther 25: 1531-43.
- Fodero-Tavoletti MT, Rowe CC, McLean CA, Leone L, Li Q-X, et al. (2009) Characterization of PiB binding to white matter in Alzheimer disease and other dementias. J Nucl Med 50: 198-204.
Citation: Brown DA, Sarkar G, Decklever TD, Curran GL, Sarkar AJ, et al. (2019) K16ApoE Enhances Aβ-associated 11C-PiB Deposition and PET Signal in APP/ PS1 Transgenic Mice. J Alzheimers Dis Parkinsonism 9: 468. DOI: 10.4172/2161-0460.1000468
Copyright: © 2019 Brown DA, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Share This Article
Recommended Journals
Open Access Journals
Article Tools
Article Usage
- Total views: 3362
- [From(publication date): 0-2019 - Apr 02, 2025]
- Breakdown by view type
- HTML page views: 2608
- PDF downloads: 754