Mini Review Open Access
Epigenetics of Brain Disorders: The Paradigm of Alzheimer's Disease
1Chairman of Genomic Medicine, Camilo Jose Cela University, Madrid, Spain
2EuroEspes Biomedical Research Center, Institute of Medical Science and Genomic Medicine, Corunna, Spain
- Corresponding Author:
- Ramon Cacabelos
EuroEspes Biomedical Research Center
Institute of Medical Science and Genomic Medicine
15165-Bergondo,Corunna, Spain
Tel: +34-981-780505;
E-mail: rcacabelos@euroespes.com
Received date: October 01, 2015; Accepted date: April 04, 2016; Published date: April 11, 2016
Citation: Cacabelos R (2016) Epigenetics of Brain Disorders: The Paradigm of Alzheimer’s Disease. J Alzheimers Dis Parkinsonism 6:229. doi: 10.4172/2161-0460.1000229
Copyright: © 2016 Cacabelos R. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Visit for more related articles at Journal of Alzheimers Disease & Parkinsonism
Abstract
Over 80% of brain disorders are associated with multiple genomic defects in conjunction with environmental factors and epigenetic phenomena. Classical epigenetic mechanisms, including DNA methylation, histone modifications, and microRNAs (miRNAs) regulation, are among the major regulatory elements that control metabolic pathways at the molecular level, with epigenetic modifications controlling gene expression transcriptionally and miRNAs suppressing gene expression post-transcriptionally. Epigenetic modifications are related to disease development, environmental exposure, drug treatment and aging. Epigenetic changes are reversible and can be potentially targeted by pharmacological intervention. Both hypermethylation and hypomethylation of DNA, chomatin changes and miRNA dysregulation are common in age-related disorders and in many neuropsychiatric, neurodevelopmental and neurodegenerative disorders. Major epigenetic mechanisms may contribute to Alzheimer’s disease (AD) pathology. Several pathogenic genes and many other AD-related susceptibility genes contain methylated CpG sites. AD brains exhibit a genome-wide decrease in DNA methylation. Pathogenic histone modifications are present in AD. Alterations in epigentically regulated miRNAs may contribute to the abnormal expression of pathogenic genes in AD. Epigenetic drugs can reverse epigenetic changes in gene expression and might open future avenues in AD therapeutics. Individual differences in drug response are associated with genetic and epigenetic variability and disease determinants. Pharmacoepigenomics deals with the influence that epigenetic alterations may exert on genes involved in the pharmacogenomic network (pathogenic, mechanistic, metabolic, transporter, and pleiotropic genes) responsible for the pharmacokinetics and pharmacodynamics of drugs (efficacy and safety), as well as the effects that drugs may have on the epigenetic machinery.
Keywords
Alzheimer’s disease; Brain disorders; Epigenetics; Epigenetic drugs; Pharmacogenomics; Pharmaco epigenomics
Introduction
Over 80% of central nervous system (CNS) disorders are polygenic/ complex disorders in which multiple defects distributed across the human genome are involved. The interaction of these pathogenic variants with diverse environmental factors and epigenetic phenomena result in the phenotypic expression of the disease [1-3]. Epigenetics involves heritable alterations of gene expression, chromatin organization, and microRNA (miRNA) regulation without changes in DNA sequence. Classical epigenetic mechanisms, including DNA methylation and histone modifications, and regulation by microRNAs (miRNAs), are among the major regulatory elements that control metabolic pathways at the molecular level, with epigenetic modifications regulating gene expression transcriptionally and miRNAs suppressing gene expression post-transcriptionally [4]. Epigenetic mechanisms are crucial to stabilize cell type-specific gene-expression programs [5]. Vertebrate genomes undergo epigenetic reprogramming during development and disease. Stable transmission of DNA methylation, transcriptomes and phenotypes from parent to clonal offspring are demonstrated in various asexual species, and clonal genotypes from natural populations show habitat-specific DNA methylation [6]. Methylation varies spatially across the genome with a majority of the methylated sitess mapping to intragenic regions [7]. Not only nuclear DNA (nDNA), but also mitochondrial DNA (mtDNA) may be subjected to epigenetic modifications related to disease development, environmental exposure, drug treatment and aging. mtDNA methylation is attracting increasing attention as a potential biomarker for the detection and diagnosis of diseases and the understanding of cellular behavior [8].
About 70% of CpG dinucleotides within the human genome are methylated. The transfer of methyl groups in CpGs is catalyzed by DNA methyl transferases (DNMT1, DNMT3A, DNMT3B).The enzymes involved in DNA de methylation include TET (ten-eleven translocation family), AID/APOBEC family, and the VER glycosylase family [9]. Histone acetylation is achieved by histone acetyltransferase (HAT); and histone deacetylation is produced by histone deacetylases (HDACs) (class I: HDAC1, 2, 3, and 8; class IIa: HDAC4, 5,7, and 9; class IIb: HDAC6 and 10; class III: SIRT1, 2, 6, 7; class IV: HDAC11) [9].
Long non-coding (lnc) RNAs are non-protein-coding RNAs, distinct from housekeeping RNAs (tRNAs, rRNAs, and snRNAs) and independent from small RNAs with specific molecular processing machinery. Over 95% of the eukaryotic genome is transcribed into non-coding RNAs and less than 5% is translated. LncRNA-mediated epigenetic regulation depends on lcnRNA interactions with proteins or genomic DNA via RNA secondary structures [10].
Epigenomic modifications are involved in a great variety of physiological and pathological conditions; of major importance are those related with major problems of health such as cardiovascular disorders, obesity, cancer, inflammatory processes, and brain disorders [11,12]. A good paradigm on the influence of epigenetic factors on human pathology is the oncogenic process in some types of cancer. For instance, myelodysplastic syndromes (MDS) are clonal diseases of the elderly characterized by chronic cytopenias, dysplasia, and a variable risk of progression to acute myeloid leukemia (AML). Aberrant methylation of tumor suppressor gene promoters has been established, suggesting that these alterations are drivers of MDS pathogenesis [13]. Epigenetic modifications are reversible and can be targeted by pharmacological intervention [14-17].
Brain disorders
Both hypermethylation and hypomethylation of DNA, chomatin changes and miRNA dysregulation are common in age-related disorders and in multiple modalities of brain disorders [9,15-17]. Altered DNA methylation patterns may account for phenotypic changes associated with human aging. Brain region-specific expression of genes can be epigenetically regulated by DNA methylation [18] and brain aging might be influenced by epigenetic changes in the neuronal microenvironment [19,20].
There are neurodevelopmental disorders in which epigenetic dysregulation plays an important role (autism spectrum disorders, Rett syndrome, fragile X syndrome, Prader-Willi syndrome, Angelman syndrome, and Kabuki syndrome [21-23]. Fragile X syndrome (FXS) is the most common monogenic form of developmental cognitive impairment with “dynamic” mutations of a CGG repeat in the 5’UTR of the FMR1 gene which is inactivated by DNA methylation and histone deacetylation [24]. Rett syndrome (RTT) is an X-linked neurodevelopmental disease caused by MECP2 mutations. The MeCP2 protein acts as a transcription repressor by binding to methylated CpG dinucleotides, and also as a transcription activator. MeCP2 is expressed in neurons and in glial cells. Reintroduction of MeCP2 into behaviorally-affected Mecp2-null mice after birth rescues neurological symptoms, indicating that epigenetic failures in RTT are reversible [25].
A growing number of congenital disorders have been linked to genomic imprinting which is caused by perturbed gene expression at one principal imprinted domain. Some imprinting disorders, including the Prader-Willi and Angelman syndromes, are caused almost exclusively by genetic mutations. In several others, including the Beckwith-Wiedemann and Silver-Russell growth syndromes, and transient neonatal diabetes mellitus, imprinted expression is perturbed mostly by epigenetic alterations at ‘imprinting control regions’ and at other specific regulatory sequences. In a minority of these patients, DNA methylation is altered at multiple imprinted loci, suggesting that common trans-acting factors are affected [26]. Maternal UPD for chromosome 7 (matUPD7) results in Silver-Russell syndrome (SRS) with typical features and growth retardation, but no gene has been conclusively implicated in SRS. Genome-scale analysis of eight matUPD7 patients, a segmental matUPD7q31-qter, a rare patUPD7 case and ten controls on the Infinium HumanMethylation450K Bead Chip with 30,017 CpG methylation probes for chromosome 7 showed highly significant clustering of DMRs only on chromosome 7, including the known imprinted loci GRB10, SGCE/PEG10, and PEG/MEST. Ten novel DMRs on chromosome 7, two DMRs for the predicted imprinted genes HOXA4 and GLI3 and one for the disputed imprinted gene PON1, and differential expression for three genes with novel DMRs, HOXA4, GLI3, and SVOP, were also demonstrated. Allele-specific expression analysis confirmed maternal only expression of SVOPL and imprinting of HOXA4 was supported by monoallelic expression. These results reported by Hannula-Jouppi et al. [27] represent the first comprehensive map of parent-of-origin specific DMRs on human chromosome 7, suggesting many new imprinted sites.
A body of novel arguments postulates the involvement of epigenetic mechanisms in the pathogenesis of autism. Mbadiwe and Millis [28] reviewed mechanisms for altering DNA-histone interactions of cell chromatin to upregulate or downregulate gene expression that could serve as epigenetic targets for therapeutic interventions. Quality of maternal care experienced during infancy is a key factor that can confer vulnerability or resilience to psychiatric disorders later in life. Experiences within an adverse caregiving environment produce aberrant DNA methylation patterns at various gene loci in the medial prefrontal cortex of developing and adult experimental animals [29].
Altered DNA methylation at the aryl hydrocarbon receptor repressor (AHRR) correlates with self-reported smoking. Smoking was associated with DNA demethylation at two distinct loci within AHRR (cg05575921 and cg21161138), and methylation status at the AHRR residue interrogated by cg05575921 was highly correlated with serum cotinine levels [30].
Epigenetic changes are also determinant in several neurodegenerative disorders such as Alzheimer’s disease, Parkinson’s disease or Huntington’s disease [9,15,16,31,32].
Alzheimer’s disease
Alzheimer’s disease (AD) is a major problem of health in developed countries. AD affects approximately 5.4 million individuals in the United States and is estimated to affect up to 16 million by 2050 [33]. In Western countries, AD is the most prevalent form of dementia (45-60%), followed by vascular dementia (VD) (30-40%), and mixed dementia (10-20%), which in people older than 85 years of age may account for over 80% of the cases. The different forms of dementia pose several challenges to our society and the scientific community: (i) they represent an epidemiological problem, and a socio-economic, psychological and family burden; (ii) most of them have an obscure/ complex pathogenesis; (iii) their diagnosis is not easy and lacks specific biomarkers; and (iv) their treatment is difficult and inefficient [2,3,31,34,35]. In terms of economic burden, approximately 10-20% of direct costs are associated with pharmacological treatment, with a gradual increase in parallel with the severity of the disease.
Pathogenesis
Our understanding of the pathophysiology of dementia, parkinsonism and other CNS disorders, has advanced dramatically during the last 30 years, especially in terms of their molecular pathogenesis and genetics. Improvement in terms of clinical outcome, however, has fallen short of expectations, in spite of efforts to identify optimal treatment regimes with one or more drugs. Potential reasons to explain this historical setback might be that: (i) the molecular pathology of dementia is still poorly understood; (ii) drug targets are inappropriate, not fitting into the real etiology of the disease; (iii) most treatments are symptomatic, but not anti-pathogenic; (iv) the genetic and epigenetic component of dementia is poorly defined; and (v) the understanding of genome-drug interactions is very limited [2,3,34-36].
In general terms, AD is a polygenic/complex disorder in which hundreds of defective genes distributed across the human genome are involved in close interaction with environmental factors, cerebrovascular dysfunction, and epigenetic changes. A growing body of information suggests that diverse epigenetic phenomena may be involved in the pathogenesis of AD; however, this field is still in a very primitive stage [9,15,16,34-38].
AD is a multifactorial, polygenic/complex disorder characterized by (i) a clinical picture with progressive memory decline, behavioral changes, and functional deterioration, (ii) neuropathological hallmarks represented by deposits of extracellular Aβ aggregates in senile plaques, cytoskeletal abnormalities with intracellular neurofibrillary tangles (NFTs) resulting from the hyperphosphorylation of the tau protein, neuronal loss, and dendritic desarborization, and (iii) a myriad of neurochemical changes and mechanistic dysfunctions which altogether conform the pathogenic phenotype of the disease. The genomic, epigenomic, proteomic, and metabolomic changes underlying these phenotypic features are candidate targets for therapeutic intervention [3].
Genomics
Genome-wide association studies (GWAS) have identified numerous disease-associated variants; however, these variants have a minor effect on disease and explain only a small amount of the heritability of this complex disorder. The search for the missing heritability has shifted attention to rare variants, copy number variants, copy neutral variants and epigenetic modifications. Over 3,000 genes distributed across the human genome have been screened for association with AD during the past 30 years [1]. In the Alzgene database [39] there are 695 genes potentially associated with AD, of which the top ten are (in decreasing order of importance): APOE (19q13.2), BIN1 (2q14), CLU (8p21-p12), ABCA7 (19p13.3), CR1 (1q32), PICALM (11q14), MS4A6A (11q12.1), CD33 (19q13.3), MS4A4E (11q12.2), and CD2AP (6p12). Potentially defective genes associated with AD represent about 1.39% (35,252.69 Kb) of the human genome, which is integrated by 36,505 genes (3,095,677.41 Kb). The highest number of AD-related defective genes is concentrated on chromosomes 10 (5.41%; 7337.83 Kb), 21 (4.76%; 2,289,15 Kb), 7 (1.62%; 2,584.26 Kb), 2 (1.56%; 3,799.67 Kb), 19 (1.45%; 854.54 Kb), 9 (1.42%; 2,010.62 Kb), 15 (1.23%; 1,264.4 Kb), 17 (1.19%; 970.16 Kb), 12 (1.17%; 1,559.9 Kb), and 6 (1.15%; 1,968.22 Kb), with the highest proportion (related to the total number of genes mapped on a single chromosome) located on chromosome 10 and the lowest on chromosome Y [3].
The genetic and epigenetic defects identified in AD can be classified into 4 major categories: Mendelian mutations, susceptibility singlenucleotide polymorphisms (SNPs), mitochondrial DNA (mtDNA) mutations, and epigenetic changes. Mendelian mutations affect genes directly linked to AD, including 32 mutations in the amyloid beta precursor protein (APP) gene (21q21)(AD1); 165 mutations in the presenilin 1 (PSEN1) gene (14q24.3)(AD3); and 12 mutations in the presenilin 2 (PSEN2) gene (1q31-q42) (AD4) [1-3,34-36,40-43]. PSEN1 and PSEN2 are important determinants of γ-secretase activity responsible for proteolytic cleavage of APP and NOTCH receptor proteins. Mendelian mutations are very rare in AD (1:1000). Mutations in exons 16 and 17 of the APP gene appear with a frequency of 0.30% and 0.78%, respectively, in AD patients. Likewise, PSEN1, PSEN2, and microtubule-associated protein Tau (MAPT) (17q21.1) mutations are present in less than 2% of the cases. Mutations in these genes confer specific phenotypic profiles to patients with dementia: amyloidogenic pathology associated with APP, PSEN1 and PSEN2 mutations, and tauopathy associated with MAPT mutations representing the two major pathogenic hypotheses for AD [1-3,34-36,40-43]. Ten novel private pathogenic copy number variations (CNVs) in 10 early-onset familial Alzheimer’s disease (EO-FAD) families overlapping a set of genes (A2BP1, ABAT, CDH2, CRMP1, DMRT1, EPHA5, EPHA6, ERMP1, EVC, EVC2, FLJ35024 and VLDLR) have also been identified [44].
Multiple polymorphic risk variants can increase neuronal vulnerability to premature death. Among these susceptibility genes, the apolipoprotein E (APOE) gene (19q13.2)(AD2) is the most prevalent as a risk factor for AD, especially in those subjects harboring the APOE-4 allele, whereas carriers of the APOE-2 allele might be protected against dementia [1,3]. Polymorphic variants in other genes (GRB-associated binding protein 2 (GAB2) [45], TLR9 rs187084 variant homozygote GG [46], LRRK2 R1628P variant [47]) might be protective, as well.
APOE-related pathogenic mechanisms are also associated with brain aging and with the neuro pathological hallmarks of AD [1-3,34,35,40-43,48-50]. mtDNA damage may also contribute to increase brain vulnerability and neuro degeneration [51,52].
Epigenomics of Alzheimer’s disease
As a complex polygenic/multifactorial disorder, in which hundreds of polymorphic variants of risk might be involved, AD fulfills the “golden rule” of complex disorders, according to which the larger the number of genetic defects distributed in the human genome, the earlier the onset of the disease and the poorer its therapeutic response to conventional treatments; and the smaller the number of pathogenic SNPs, the later the onset of the disease, and the better the therapeutic response to different pharmacological interventions [1,3,48,50,53]; however, conventional genomics do not explain in full AD pathogenesis in which epigenetics may help to understand some enigmatic events. DNA methylation, histone modifications and chromatin remodeling and non-coding RNA dysregulation may contribute to AD pathology, although evidence is still very limited [9,15,16,54-56]. Pharmaceuticals, pesticides, air pollutants, industrial chemicals, heavy metals, hormones, nutrition, and behavior can change gene expression through a broad array of gene regulatory mechanisms. Mechanisms include regulation of gene translocation, histone modifications, DNA methylation, DNA repair, transcription, RNA stability, alternative RNA splicing, protein degradation, gene copy number, and transposon activation [57]. Genetic variation associated with different diseases interferes with microRNA-mediated regulation by creating, destroying, or modifying miRNA binding sites. miRNA-target variability is a ubiquitous phenomenon in the adult human brain, which may influence gene expression in physiological and pathological conditions. One of the major roles of lncRNAs in the nucleus is the regulation of gene expression at the transcriptional level via histone or DNA modification [58]. Epigenetic mechanisms and miRNAs have recently been shown to closely interact with each other, thereby creating reciprocal regulatory circuits, which appear to be disrupted in AD [59]. Brain hypoperfusionrelated changes in DNA methylation may also contribute to accelerate neuronal death. Short-term, sub-lethal hypoxia results in long-lasting changes to genome-wide DNA methylation status, and some of these changes can be highly correlated with transcriptional modulation in a number of genes involved in functional pathways [60]. Inflammatory mechanisms contribute substantially to secondary tissue injury after brain ischemia. Regulatory T cells (RTC) are endogenous modulators of postischemic neuroinflammation. HDACi, using trichostatin A, increases the number of RTC, boosts their immunosuppressive capacity and interleukin (IL)-10 expression, reduces infarct volumes and behavioral deficits after cortical brain ischemia, attenuates cerebral proinflammatory cytokine expression, and increases the number of brain-invading RTC. A similar effect is obtained using tubastatin, a specific inhibitor of HDAC6 and a key HDAC in Foxp3 regulation. The neuroprotective effect of HDACi depends on the presence of Foxp3+ RTC, and in vivo and in vitro studies show that the anti-inflammatory cytokine IL-10 was their main mediator [61].
Memory decline is a seminal symptom in dementia. Gene expression is required for long-lasting forms of memory. Epigenetic mechanisms do not only provide complexity in the protein regulatory complexes that control coordinate transcription for specific cell function, but the epigenome encodes critical information that integrates experience and cellular history for specific cell functions as well. Epigenetic mechanisms provide a unique mechanism of gene expression regulation for memory processes. Negative regulators of gene expression, such as HDACs, have powerful effects on the formation and persistence of memory. HDAC inhibition transforms a subthreshold learning event into robust long-term memory and generates a form of long-term memory that persists beyond the point at which normal long-term memory fails [62]. Whereas increments in histone acetylation have consistently been shown to favor learning and memory, a lack thereof has been causally implicated in cognitive impairments in neurodevelopmental disorders, neurodegeneration and aging. As histone acetylation and cognitive functions can be pharmacologically restored by histone deacetylase inhibitors, this epigenetic modification might constitute a molecular memory aid on the chromatin and a new template for therapeutic interventions against cognitive decline [63].
DNA methylation
DNA methylation is involved in memory processes: (i) hippocampal DNMT expression is up-regulated during consolidation of contextual fear memory; (ii) intra-hippocampal administration of DNMT inhibitors blocks this memory consolidation, and DNMTiinduced memory deficits can be overcome by pretreatment with HDAC inhibitors; (iii) rapid changes in DNA methylation at the time of learning provide bi-directional transcriptional regulation of memory promoting and suppressing genes; (iv) conditional knockout mice lacking both DNMT1 and DNMT3a forebrain expression display memory dysfunction and deficits in long-term plasticity in the hippocampus; (v) hippocampal learning triggers gene-specific hypermethylation in the cortex which persists for weeks; and (vi) DNA methylation can be both dynamic, supporting synaptic consolidation, and stable, supporting system consolidation [64].
Several pathogenic genes (APP, PS1, APOE, BACE, CLU) and many other AD-related susceptibility genes contain methylated CpG sites and a genome-wide decrease in DNA methylation has been reported in AD [9,55] (Table 1). The promoter region of the APP gene is hypomethylated, this contibuting to a potential enhancement of Aβ production; however, some authors have reported no relevant changes in APP methylation, with an epigenetic drift in AD samples [65]. Methylation status of repetitive elements (i.e. Alu, LINE-1 and SAT-α) is a major contributor of global DNA methylation patterns. The study of global DNA methylation levels for long interspersed nuclear element 1 (LINE-1) repetitive sequences in patients with AD and controls did not provide clear results. In one study, no differences in LINE-1 methylation levels were found between patients and controls [66]; whereas in another study, LINE-1 methylation was found increased in AD patients compared with healthy volunteers [67]. In AD, both hypomethylation and hypermethylation of specific genes have been reported [9] (Table 1). DNA methylation of the APP promoter was found to be decreased in the brain of autopsy cases older than 70 years of age as compared with younger cases [68]. The intracellular domain of APP (AICD) has emerged as a key epigenetic regulator of gene expression controlling a diverse range of genes, including APP itself, the amyloid-degrading enzyme neprilysin, and aquaporin-1 [69]. Abnormal processing of neuronal cell membrane APP is accompanied by elevated human serum and CSF levels of 24-hydroxycholesterol, an endogenous ligand of Liver X receptor (LXR-α). There is an epigenomic pathway that connects LXR-α activation with genes involved in the regulation of aberrant Aβ production, leading to the generation of toxic and inflammatory mediators responsible for neuronal death. LXR-α activation by its specific endogenous or exogenous ligands results in the overexpression of the PAR-4 gene and suppression of the AATF gene through its inherent capacity to regulate genes coding for SREBP and NF-κB. Overexpression of the PAR-4 gene is accompanied by aberrant Aβ production followed by ROS generation and subsequent neuronal death. Aβ-induced heme oxygenase-1 can ensure cholesterol-oxidation to provide endogenous ligands for the sustained activation of neuronal LXR-α-dependent epigenomic pathways, leading to neuronal death in AD [70].
| Pathogenic genes | Locus | Promoter length (bp) | 3'UTR length | Defective protein | Methylation |
|---|---|---|---|---|---|
| APOE apolipoprotein E | 19q13.32 | 996 | -- | apolipoprotein E | Hypomethylated |
| APP amyloid beta (A4) precursor protein |
21q21.3 | 1086 | 1176 | amyloid beta (A4) protein | Hypomethylated |
| BACE1 beta site APP cleaving enzyme 1 |
11q23.2-q23.3 | 987 | 3994 | beta-secretase 1 | Hypomethylated |
| CREB cAMP response element binding protein 1 |
2q33.3 | 1026 | -- | cAMP response element binding protein 1 | Hypomethylated |
| MAPT microtubule-associated protein tau | 17q21.31 | 1094 | -- | microtubule-associated protein tau | Hypermethylated |
| MTHFR methylene Tetrahydropholate reductase | 1p36.22 | 959 | -- | methylene tetrahydropholate reductase | Hypermethylated |
| NCSTN nicastrin | 1q22-q23 | 922 | 766 | nicastrin | Hypermethylated |
| MME Membrane metallo- endopeptidase | 3q25.1-q25.2 | 972 | 3330 | neprilysin | Hypermethylated |
| PP2A protein phosphatase 2 | 9q34 | 981 | 1598 | serine/threonine-protein phosphatase 2A activator | Hypomethylated |
| PSEN1 presenilin 1 |
14q24.2 | 929 | 1198 | presenilin 1 | Hypomethylated |
| S100A2 S100 calcium binding protein A2 |
1q21.3 | 902 | 400 | protein S100-A2 | Hypomethylated |
| SORBS3 sorbin and SH3 domain containing 3 |
8p21.3 | 972 | 939 | vinexin | Hypermethylated |
| SPTBN4 spectrin beta nonerythrocytic 4 | 19q13.13 | 947 | 993 | spectrin beta chain, non-erythrocytic 4 | Hypermethylated |
| TBXA2R thromboxane A2 receptor | 19p13.3 | 983 | 1335 | thromboxane A2 receptor | Hypermethylated |
| TMEM59 transmembrane protein 59 | 1p32.3 | 1016 | 628 | transmembrane protein 59 | Hypomethylated |
Table 1: Gene methylation patterns in Alzheimer’s disease.
Hyper phosphorylated tau is responsible for the formation of NFTs. Changes in methylation status differ among transcription factor binding sites of tau promoter. Binding sites for GCF (granulocyte chemotactic factor), responsible for repression of GC-rich promoters, were found to be hypo methylated, whereas binding sites for the transcriptional activator SP1 (specificity factor 1) were hyper methylated [71]. High levels of Hcy may induce tau hyper phosphorylation, NFT formation, and SP formation via inhibition of methyl transferases and hypomethylation of protein phosphatase 2A (PP2A), a dephosphorylating enzyme of phosphorylated tau [72].
Histone modifications
Histone modifications are present in AD [9,15,16,63,73]: (i) histone acetylation is reduced in AD brain tissues [74] and in AD transgenic models [63]; (ii) levels of HDAC6, a tau-interacting protein and a potential modulator of tau phosphorylation and accumulation, are increased in cortical and hippocampal regions in AD [75]; (iii) SIRT1 is decreased in the parietal cortex of AD patients, and the accumulation of Aβ and tau in AD brains might be related to the loss of SIRT1 [76], since SIRT1 may reduce Aβ production, activating the transcription of ADAM10 [77]; (iv) in the brains of twins discordant for AD, tri methylation of H3K9, a marker of gene silencing, and condensation of heterochromatin structure, are increased in the temporal cortex and hippocampus of the AD twin as compared to the twin devoid of AD neuropathology [78]; (v) phosphorylation of H3S10, a key regulator in chromatin compaction during cell division, is increased in the cytoplasm of hippocampal neurons in AD cases [79]; (vi) evidence of DNA damage, as reflected by phosphorylated H2AX at Ser139, is present in hippocampal astrocytes of AD patients [80]; (vii) long-term potentiation (LTP) and memory deficits in APP/PS1 transgenic mice might be mediated in part by decreased H4 acetylation; improving histone acetylation level restores learning after synaptic dysfunction [81]; (viii) acetylation of H3 and H4 is increased in 3xTg-AD neurons relative to non-transgenic neurons [82]; (ix) nuclear translocation of EP300 interacting inhibitor of differentiation 1 (EID1), a CBP/p300 inhibitory protein, is increased in the cortical neurons of AD patients, and overexpression of EID1 is reported to reduce hippocampal LTP and to impair cognitive function via inhibiting CBP/p300 acetyl trasferase activity and disrupting neuronal structure [83]; (x) memory formation leads to a transient increase in acetylation on lysine residues within H2B, H3, H4 [84,85]; (xi) inhibition of HDAC induces dendritic sprouting, increases synaptic number, and improves long-term memory [86]; (xii) overexpression of neuronal HDAC2 decreases dendritic spine density, synapse number, synaptic plasticity and memory formation, and HDAC2 deficiency increases synapse number and memory facilitation [87,88]; (xiii) HDAC4 is involved in learning and synaptic plasticity, and selective inhibition of HDAC4 activity may deteriorate learning and memory [89]; (xiv) treatment of hippocampal neurons with HDAC inhibitors facilitates Bdnf expression via hyper acetylation of histones at the Bdnf promoters [90,91]; (xv) histone(H3K4) methylation participates in the regulation of Bdnf expression and memory formation [92]; (xvi) histone methylation also facilitates memory consolidation coupled with histone acetylation; inhibition of HDACs with sodium butyrate (NaB) causes an increase in H3K4 trimethylation and a decrease in H3K9 dimethylation in the hippocampus after fear conditioning [92]; (xvii) histone H3 acetylation, methylation and phosphorylation is increased in the prefrontal cortex of Tg2576 mice, and histone H4 acetylation is increased in the hippocampal CA1 neurons of these transgenic mice [15,16,93].
Non-coding RNAs
miRNAs belong to the class of non-coding regulatory RNA molecules of ~22 nt length and are now recognized to regulate ~60% of all known genes through post-transcriptional gene silencing (RNA interference)(RNAi). Alterations in epigentically regulated miRNAs may contribute to the abnormal expression of pathogenic genes in AD [59,94]. Several lncRNAs are dysregulated in AD (Sox2OT, 1810014B01Rik, BC200, BACE1-AS, NAT-Rad18, 17A, GDNFOS), Parkinson’s disease (naPINK1, Sox2OT, 1810014B01Rik, BC200), and Huntington’s disease (HAR1F, HTTAS, DGCR5, NEAT1, TUG1) [94]. Examples of miRNAs directly linked to AD pathogenesis include miR-34a (1p36.22), miR-34b/c (11q23.1), miR-107 (10q23.31), miR- 124 (8p23.1/8p12.3/20q13.33), miR-125b (11q24.1/21q21.1), and miR-137 (1p21.3); and examples of epigenetically regulated miRNAs with targets linked to AD pathogenesis are let-7b (22q13.1), miR-9 (1q22/5q14.3/15q26.1), miR-132/212 (17p13.3), miR-146a (5q34), miR- 148a (7p15.2), miR-184 (15q25.1), and miR-200 (miR-200b/200a/429, 1p36.33; miR-200c/141, 12p13.31) [59].
miRNAs can be used as biomarkers to discriminate different disease forms, staging and progression, as well as prognosis [95]. A unique circulating 7-miRNA signature (hsa-let-7d-5p, hsa-let-7g-5p, hsa-miR- 15b-5p, hsa-miR-142-3p, hsa-miR-191-5p, hsa-miR-301a-3p and hsamiR- 545-3p) reported by Kumar et al. [95] in plasma, could distinguish AD patients from normal controls with >95% accuracy. Leidinger et al. [96] showed a novel miRNA-based signature for detecting AD from blood samples. Using this 12-miRNA signature, they differentiated between AD and controls with an accuracy of 93%, a specificity of 95% and a sensitivity of 92%. The differentiation of AD from other neurological diseases (MCI, multiple sclerosis, Parkinson disease, major depression, bipolar disorder and schizophrenia) was possible with accuracies between 74% and 78%. Alexandrov et al. [97] found increased levels of miRNA-9, miRNA-125b, miRNA-146a, miRNA-155 in the CSF and brain tissue-derived extracellular fluid from patients with AD, suggesting that these miRNAs might be involved in the modulation or proliferation of miRNA-triggered pathogenic signaling in AD brains.
AD-related SNPs interfere with miRNA gene regulation and affect AD susceptibility. The significant interactions include target SNPs present in seven genes related to AD prognosis with the miRNAs- miR- 214, -23a & -23b, -486-3p, -30e*, -143, -128, -27a &-27b, -324-5p and -422a. The dysregulated miRNA network contributes to the aberrant gene expression in AD [37,38,98].
Epigenetic Drugs
Epigenetic drugs (Tables 2 and 3) reverse epigenetic changes in gene expression and might open future avenues in AD therapeutics and other major problems of health [9,15,16,73,99-105]. Epigenetic effects are exerted by a variety of factors and evidence increases that common drugs may induce alterations in DNA methylation patterns or histone conformations. These effects occur via chemical structural interactions with epigenetic enzymes, through interactions with DNA repair mechanisms [106,107]. Several inhibitors of histone deacetylation and DNA methylation have been approved by the US FDA for hematological malignancies [15-17,99] (Table 2).
| DNA methyltransferase inhibitors Nucleoside analogs 5-Aza-2’-deoxycytidine (Decitabine) 5-Azacytidine (Azacitidine) Small molecules Hydralazine Procainamide RG108 [2-(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)-3-(1H-indol-3-yl)propanoic acid]) Natural products Curcumin derivatives RG-108 SGI-1027 Psammaplins Tea polyphenols Epigallocatechin-3-gallate Catechins Catechin Epicatechin Bioflavonoids Quercetin Genistein Fisetin Antisense oligonucleotide inhibitors ncRNAs (miRNAs) Histone deacetylase (HDAC) inhibitors Short-chain fatty acids Sodium butyrate Sodium phenyl butyrate Valproic acid Pivaloyloxymethyl butyrate (AN-9, Pivanex) Hydroxamic acids Suberoylanilide hydroxamic acid (SAHA, Vorinostat) Oxamflatin Pyroxamide Trichostatin A (TSA) m-Carboxycinnamic acid bis-hydroxamide (CBHA) Derivatives of the marine sponge Psammaplysilla purpurea NVP-LAQ824 NVP-LBH589 LBH-589 (Panobinostat) ITF2357 (Givinostat) PXD101 (Belinostat) JHJ-26481585 CHR-3996 CHR-2845 PCI-24781 Cyclic peptides Romidepsin (Depsipeptide, FR901228) < Apicidin Cyclic hydroxamic acid-containing peptides (CHAPS) Trapoxin A and B Chlamydocin HC toxin Bacterial FK228 Benzamides MS-275 (Entinostat) CI-994 RGFP136 MGCD0103 (Mocetinostat) Compound 60 Ketones Trifluoromethyl ketone Sirtuin modulators Sirtuin inhibitors Nicotinamide/niacinamide Suramin AGK-2 Sirtinol Salermide MS3 Splitomycin Cambiol SEN-196 Dihydrocoumarin Tenovin UVI5008 Sirtuin activators Resveratrol SRT-501 SRT-1460 SRT-1720 SRT-2183 GSK-184072 Quercetin Piceatannol Miscellaneous compounds 3-Deazaneplanocin A (DZNep) Tubacin EVP-0334 6-([18F]Fluoroacetamido)-1-hexanoicanilide Quinazolin-4-one derivatives (E)-3-(2-Ethyl-7-fluoro-4-oxo-3-phenethyl-3,4-dihydroquinazolin-6-yl)-N-hydroxyacrylamide N-Hydroxy-3-(2-methyl-4-oxo-3-phenethyl-3,4-dihydro-quinazolin-7-yl)-acrylamide Histone acetyltransferase modulators Histone acetyltransferase inhibitors Curcumin (Diferuloylmethane) Lys-CoA H3-CoA-20 Anacardic acid Garcinol Histone aceyltransferase activators N-(4-Chloro-3-trifluoromethyl-phenyl)-2-ethoxy-6-pentadecyl-benzamide Pentadecylidenemalonate 1b (SPV-106) Histone methyltransferase inhibitors Lysine methyltransferase inhibitors S-Adenosylmethionine (SAMe) SAMe analogs Chaetocin BIX-01294 BIX-01338 UNC0224 EZH2 (KMT6) inhibitors Deazaneplanocin A Arginine methyltransferase inhibitors AMI-1 Histone demethylase inhibitors Lysine-specific demethylase 1 (LSD1) inhibitors Tranylcypromine Parnate (S)-4-(2-(5-(Dimethylamino)naphthalene-1-sulfonamido)-2-phenylacetamido)-N-hydroxybenzamide (D17) Non-coding RNAs miRNAs RNAi Other potential epigenetic treatments Small molecule inhibitors to chromatin-associated proteins DOT1L histone methyltransferase inhibitors EPZ004777 EPZ-5676 SGC0946 EZH2 histone methyltransferase inhibitors GSK126 GSK343 EPZ005687 EPZ-6438 EI1 UNC1999 G9A histone methyltransferase inhibitors BIX1294 UNC0321 UNC0638 NC0642 BRD4770 PRMT3 histone methyltransferase inhibitors 14u PRMT4 (CARM1) histone methyltranferase inhibitors 17b MethylGene LSD1 histone demethylase inhibitors Tranylcypromine ORY-1001 BET histone demethylase inhibitors JQ1 IBET762 IBET151 PFI-1 BAZ2B histone demethylase inhibitors GSK2801 L3MBTL1 chromodomain inhibitors UNC669 L3MBTL3 chromodomain UNC1215 Bromodomain inhibitors LP99 RVX-208 Chromodomain inhibitors |
Source: Adapted from Cacabelos (ref. 17) and Cacabelos and Torrellas (Ref. 15).
Table 2: Classification of selected epigenetic drugs.
Reducing the hyper methylation levels in some pathogenic genes may be an alternative therapy in AD, in addition to conventional treatments (cholinesterase inhibitors: donepezil, rivastigmine, galantamine; NMDA partial antagonists: memantine) (Table 4) or novel therapies (immune therapy/vaccination; secretase inhibitors; Aβ breakers; other unconventional treatments) [3]. Examples of DNMT inhibitors include (i) nucleoside analogs (5-aza-2’-deoxycytidine (Decitabine), 5-azacytidine (Azacitidine)), (ii) small molecules (hydralazine, procainamide, RG108 [2-(1,3-dioxo-1,3-dihydro 2H-isoindol-2-yl)-3-(1H-indol-3-yl)propanoic acid]), (iii) natural products (curcumin derivatives (RG-108, SGI-1027), psammaplins, tea polyphenols (epigallocatechin-3-gallate), catechins (catechin, epicatechin), bioflavonoids (quercetin, genistein, fisetin)), (iv) antisense oligonucleotide inhibitors (MG98), and (v) ncRNAs (miRNAs) [11,34,35] (Tables 2 and 3).
| Drug | Properties | Pharmacogenetics |
|---|---|---|
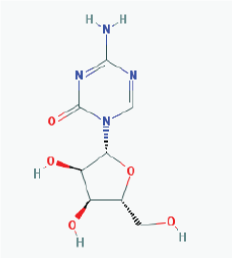 |
Name: 5-Azacytidine, Azacitidine, Azacytidine, Ladakamycin, Vidaza, Mylosar, Azacitidinum, 5-AZAC IUPAC Name: 4-Amino-1-[(2R,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-1,3,5-triazin-2-one Molecular Formula: C8H12N4O5 Molecular Weight: 244.20468 Category: Pyrimidine nucleoside cytidine analog Mechanism: DNA methyltransferase inhibitor, Telomerase inhibitor -Target:DNA (cytosine-5)-methyltransferase 1 (DNMT1) -Interactions:Cytidine deaminase Effect: Antineoplastic, Antimetabolite.Methylates CpG residues. Methylates hemimethylated DNA. Mediates transcriptional repression by direct binding to HDAC2 |
Pathogenic genes: ALDH3A1, CDKN2A, MGMT, PLA2R1, RRM1, TNFRSF1B Mechanistic genes: ALDH1A1, DAPK1, DNMT1, DPYD, CDKN2A, MGMT, PLCB1 Metabolic genes: Substrate: CDA, DCK, SLC28A1, SLC29A1, RRM1, RRM2, UCK1, UCK2 Inhibitor: CYP1A2 (weak), CYP2E1 (weak), DNMT1 Inducer: SULT1C2 Transporter genes: SLC5A5,SLC28A1, SLC29A1 Pleiotropic genes: BLK |
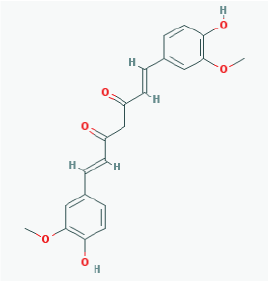 |
Name: Curcumin, Diferuloylmethane, Natural yellow 3, Turmeric yellow, Turmeric, Kacha haldi, Gelbwurz, Curcuma, Haldar, Souchet IUPAC Name: (1E,6E)-1,7-bis(4-hydroxy-3-methoxyphenyl)hepta-1,6-diene-3,5-dione Molecular Formula: C21H20O6 Molecular Weight: 368.3799 Category: Natural product (Curcuma longa) Mechanism:Histone acetyltransferase (HAT) inhibitor Effect: Non-steroidal anti-inflammatory agent; Antineoplastic; Antioxidant; Cognitive enhancer; Coloring agent; Enzyme inhibitor |
Pathogenic genes: BACE1, CCND1, CDH1, GSK3B, IL1A, IL6, JUN, MSR1, PSEN1, PTGS2, SNCA, SREBF1, TNF Mechanistic genes: AKT1,PRKAs, BACE1, CCND1, CDH1, CDKs, CRM1, CTNNB1, EGF, GSK3B, HDACs, HIF1A, IL1A, IL6, JUN, MMPs, MSR1, NFKB1, NOS2, PDGFRs, PSEN1, PTGS2, SNCA, SOCS1, SOCS3, SREBF1, STAT3, TNF, VEGFA Metabolic genes: Inhibitor: CYP2C8, CYP2C9, EP300 Inducer: CYP2C8, CYP2C9, CYP2D6, CYP3A4 Transporter genes: ABCA1, SNCA Pleiotropic genes: CTNNB1, MSR1 |
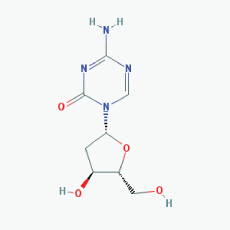 |
Name:Decitabine, 5-Aza-2'-deoxycytidine, Dacogen, Dezocitidine, 2'-Deoxy-5-azacytidine IUPAC Name: 4-Amino-1-[(2R,4S,5R)-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-1,3, 5-triazin-2-one Molecular Formula: C8H12N4O4 Molecular Weight: 228.20528 Category: Nucleoside Mechanism: DNA methyltransferase inhibitor -Target:DNA (cytosine-5)-methyltransferase 1 (DNMT1) -Interactions:Deoxycytidine kinase Effect: Antineoplastic, Antimetabolite, Enzyme inhibitor, Teratogen |
Pathogenic genes: BRCA1, CDKN2B, DNMT3A, EGFR, FOS, MGMT, MLH1, MMP9, MYC, NOS3, NQO1, TP53, VHL Mechanistic genes: APAF1, BRCA1, CDKN2B, EGFR, ICAM1, MGMT, MLH1, MMP2, MMP9, MYC, NOS3, TIMP3, TP53, VHL Metabolic genes: Substrate: DCK, DNMT1, CDA, SLC29A1 Inhibitor: DNMT1, DNMT3B Inducer: DPYD Transporter genes: ABCs, SLC15s, SLC22s, SLC28A1, SLC29As Pleiotropic genes: HBG1, NQO1, NTRK2, MMP2, MSH2 |
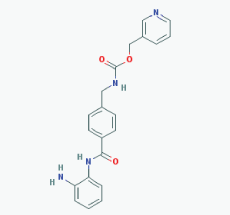 |
Name: Entinostat, ms-275, 209783-80-2, SNDX-275, MS 275, MS-27-275, SNDX 275, Histone Deacetylase Inhibitor I, S1053_Selleck, MS 27-275 IUPAC Name: Pyridin-3-ylmethyl N-[[4-[(2-aminophenyl)carbamoyl]phenyl]methyl]carbamate Molecular Formula: C21H20N4O3 Molecular Weight: 376.4085 Category: Benzamide Mechanism: Class I HDAC inhibitor (HDAC1, 2, 3) Effect: Antineoplastic agent; Histone deacetylase inhibitor; Memory enhancer |
Pathogenic genes: CDH1 Mechanistic genes: CDH1, HDAC1, HDAC2, HDAC3, KLRK1 Metabolic genes: Inhibitor: HDAC1, HDAC2, HDAC3 Inducer: CYP19A1 |
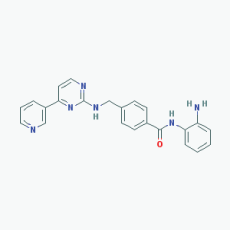 |
Name:Mocetinostat, MGCD0103, 726169-73-9, MGCD-0103, MGCD 0103, N-(2-Aminophenyl)-4-([[4-(pyridin-3-yl)pyrimidin-2 yl]amino]methyl)benzamide IUPAC Name: N-(2-Aminophenyl)-4-[[(4-pyridin-3-ylpyrimidin-2-yl)amino]methyl] benzamide Molecular Formula: C23H20N6O Molecular Weight: 396.4445 Category: Benzamide Mechanism: Class I HDAC inhibitor (HDAC1, 2, 3); Class IV HDAC inhibitor (HDAC11) Effect: Antineoplastic agent; Histone deacetylase inhibitor |
Pathogenic genes: CDKN1A, CDKN2B, TNF Mechanistic genes: CDKN1A, CDKN2B, HDAC1, HDAC2, HDAC3, HDAC11, NFKB2, TNF Metabolic genes: Inhibitor: HDAC1, HDAC2, HDAC3,HDAC11 |
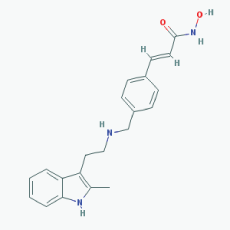 |
Name: Panobinostat,LBH-589, 404950-80-7, LBH589, Faridak, NVP-LBH589, LBH 589, S1030_Selleck, AC1OCFY8, Panobinostat (LBH589) IUPAC Name: (E)-N-hydroxy-3-[4-[[2-(2-methyl-1H-indol-3-yl)ethylamino]methyl]phenyl] prop-2-enamide Molecular Formula: C21H23N3O2 Molecular Weight: 349.42622 Category: Hydroxamic acid Mechanism: Class I HDAC inhibitor (HDAC1, 2, 3, 8); Class IIa HDAC inhibitor (HDAC4, 5, 7, 9); Class IIb HDAC inhibitor (HDAC6, 10); Class IV HDAC inhibitor (HDAC11); Pan-histone deacetylase inhibitor Effect: Antineoplastic agent; Histone deacetylase inhibitor |
Pathogenic genes: CDKN1A, EGFR, IL6, RASSF1 Mechanistic genes: AKT1, CDKN1A, DAPK1, DNMT1, EGFR, HDACs, HIST3H3, HIST4H4, HSP90As, IL6, IL10, IL12, IL23A, NFKB2, RASSF1,TLR3 Metabolic genes: Substrate: CYP2C19, CYP2D6, CYP3A4 Inhibitor: AKT1, CYP19A1 (strong), HDACs Pleiotropic genes: IL10 |
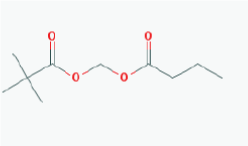 |
Name: Pivanex, AN-9, Pivalyloxymethyl butyrate, AN 9, 122110-53-6, BRN 4861411, ((2,2 Dimethylpropanoyl)oxy)methyl butanoate IUPAC Name: Butanoyloxymethyl 2,2-dimethylpropanoate Molecular Formula: C10H18O4 Molecular Weight: 202.24752 Category: Short-chain fatty acid Mechanism: Class I HDAC inhibitor (HDAC1, 2, 3, 8) Effect: Antineoplastic agent; Histone deacetylase inhibitor |
Pathogenic genes: BCL2, TP53 Mechanistic genes: BAX, BCL2, BCR-ABL, HDACs, TP53 Metabolic genes: Inhibitor: ABCB1, HDACs Transporter genes: ABCB1 |
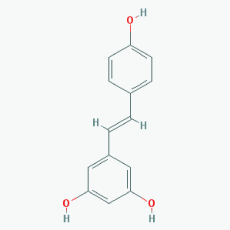 |
Name: Resveratrol, trans-resveratrol, 501-36-0, 3,4',5-Trihydroxystilbene, 3,4',5-Stilbenetriol, 3,5,4'-Trihydroxystilbene, Resvida, (E)-resveratrol IUPAC Name: 5-[(E)-2-(4-Hydroxyphenyl)ethenyl]benzene-1,3-diol Molecular Formula: C14H12O3 Molecular Weight: 228.24328 Category: Natural polyphenol Mechanism:SIRT1 inducer/activator Effect: Non-steroidal antiinflammatory agent; Anticarcinogenic; Antimutagenic; Antineoplastic; Antioxidant; Platelet aggregation inhibitor; Enzyme inhibitor; Lifespan extension; Memory improvement; Aβ decrease; Reduction of plaque formation |
Pathogenic genes: BCL2, CAV1, ESR1, ESR2, GRIN2B, NOS3, PTGS2, TNFRSF10A, TNFRSF10B Mechanistic genes: APP, ATF3, BAX, BAK1, BBC3, BCL2, BCL2L1, BCL2L11, BIRC5, CASP3, CAV1, CFTR, ESR1, ESR2, GRIN1, GRIN2B, HTR3A, NFKB1, NOS3, PMAIP1, PTGS1, PTGS2, SIRT1, SIRT3, SIRT5, SRC, TNFRSF10A, TNFRSF10B, TRPs Metabolic genes: Substrate: CYP1A1, CYP1A2, CYP1B1, CYP2E1, GSTP1, PTGS1, PTGS2 Inhibitor: CYP1A1, CYP1B1, CYP2C9, CYP2D6, CYP3A4, NQO2 Inducer: CYP1A2, SIRT1 Transporter genes: ABCC1, ABCC2, ABCC3, ABCC4, ABCC8, ABCG1, ABCG2, CFTR, TRPs |
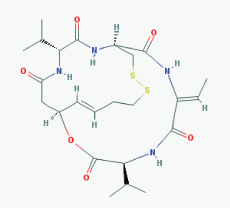 |
Name: Romidepsin, Depsipeptide, Chromadax, Istodax, Antibiotic FR 901228, FK228, FR 901228, FK-228, NSC 630176, NSC-630176 IUPAC Name: (1S,4S,7Z,10S,16E,21R)-7-ethylidene-4,21-di(propan-2-yl)-2-oxa-12, 13-dithia-5,8,20,23-tetrazabicyclo[8.7.6]tricos-16-ene-3,6,9,19, 22-pentone Molecular Formula: C24H36N4O6S2 Molecular Weight: 540.69584 Category: Cyclic peptide Mechanism:Class I HDAC inhibitor (HDAC1, 2, 3, 8); Class IIa HDAC inhibitor (HDAC4,5,7,9); Class IIb HDAC inhibitor (HDAC6, 10); Class IV HDAC inhibitor (HDAC11) Effect: Antibiotic; Antineoplastic agent; Histone deacetylase inhibitor |
Pathogenic genes: BCL2, CCDN1, CDKN1A, MYC, NF2, RB1, ROS1, TNFSF10, VHL Mechanistic genes: BCL2, CCDN1, CDKN1A, FLT1, HDAC1, HDAC2, HDAC3, HDAC4, HSP90As, KDR, MYC, NF2, TNFSF10, VEGFs, VHL Metabolic genes: Substrate: ABCB1, ABCG2, CYP1A1 (minor), CYP2B6 (minor), CYP2C19 (minor), CYP3A4 (major), CYP3A5 (minor), NR1I3, SLCO1B3 Inhibitor: ABCB1, HDACs Inducer: ABCG2 Transporter genes: ABCB1, ABCC1, ABCG2, SLCO1B3 Pleiotropic genes: CDH1, CDKN1A |
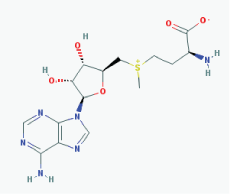 |
Name: S-Adenosylmethionine, Ademetionine, AdoMet, Donamet, S-adenosyl-L-methionine, SAMe, Methioninyladenylate, SAM-e, adenosylmethionine IUPAC Name: (2S)-2-Amino-4-[[(2S,3S,4R,5R)-5-(6-aminopurin-9-yl)-3,4-dihydroxyoxolan-2-yl]methyl-methylsulfonio]butanoate Molecular Formula: C15H22N6O5S Molecular Weight: 398.43738 Category: Methyl radical donor Mechanism:Histone methyltransferase inhibitor Effect: Antineoplastic; Antiinflammatory; Memory enhancer; PSEN1 repressor |
Pathogenic genes: AKT1, ERK, GNMT, MAT1A, PSEN1 Mechanistic genes: AMD1, CAT, CBS, GCLC, GNMT, GSS, NOS2, ROS1, STAT1, TNF Metabolic genes: Substrate: COMT, GNMT, TPMT, SRM Inhibitor: ABCB1, CYP2E1, NOS2 Transporter genes: SLC25A26 Pleiotropic genes: CAT, TNF |
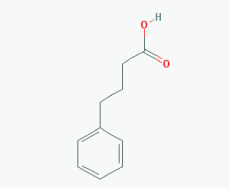 |
Name: Sodium phenylbutyrate,Buphenyl, 4-Phenylbutiric acid, 4-Phenylbutanoic acid, Benzenebutanoic acid, Benzenebutyric acid, Butyric acid, 4-phenyl-, 1821-12-1, gamma-Phenylbutyric acid, IUPAC Name: 4-Phenylbutanoic acid Molecular Formula: C10H12O2 Molecular Weight: 164.20108 Category: Short-chain fatty acid Mechanism: Class I HDAC inhibitor (HDAC1, 2, 3, 8); Class IIa inhibitor (HDAC4,5,7,9); Class IIb inhibitor (HDAC6,10) Effect: Antineoplastic agent; Histone deacetylase inhibitor; Memory improvement; pTau decrease via GSK3β inactivation; C99 and Aβ decrease; Amyloid burden reduction |
Pathogenic genes: ARG1, ASS1, BCL2, CPS1, NAGS, OTC Mechanistic genes: BCL2, BDNF, EDN1, HDACs, HSPA8, ICAM1, NFKB2, NT3, VCAM1 Metabolic genes: Inhibitor: HDACs Inducer: ARG1, CFTR, CYP2B6, NFKB2 Transporter genes: CFTR Pleiotropic genes: ASL, BDNF, VCAM1 |
 |
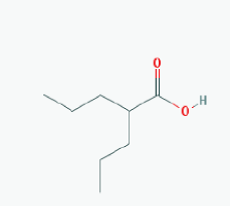
Name: Suramin, Naphuride, Germanin, Naganol, Belganyl, Fourneau, Farma, Antrypol, Suramine, Naganin IUPAC Name: 8-[[4-methyl-3-[[3-[[3-[[2-methyl-5-[(4,6, 8-trisulfonaphthalen-1-yl)carbamoyl]phenyl]carbamoyl]phenyl] carbamoylamino]benzoyl]amino]benzoyl]amino]naphthalene-1,3,5-trisulfonic acid Molecular Formula: C51H40N6O23S6 Molecular Weight: 1297.2797 Category: Polyanionic compound Mechanism: Class III HDAC/Sirtuin inhibitor (SIRT1-3) Effect:Antineoplastic Agent; Trypanocidal Agent; Antiparasitic; Antinematodal (African trypanosomiasis, Onchocerca); Sirtuin inhibitor |
Mechanistic genes: FSHR, IL10, P2RY2, PDGFRB, RYR1, SIRT1,SIRT2, SIRT3, SIRT5 Metabolic genes: Inhibitor: SIRT1, SIRT2, SIRT3 |
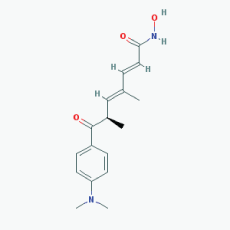 |
Name:Trichostatin A,58880-19-6, TSA, Trichostatin A (TSA), CHEBI:46024, TSA; 2,4-Heptadienamide, 7-(4-(dimethylamino)phenyl)-N-hydroxy-4,6-dimethyl-7-oxo- 7-(4-(Dimethylamino)phenyl)-N-hydroxy-4,6-dimethyl-7-oxo-2,4-heptadienamide; [R-(E,E)]-7-[4-(Dimethylamino)phenyl]-N-hydroxy-4,6-dimethyl-7-oxo-2,4-heptadienamide IUPAC Name: (2E,4E,6R)-7-[4-(dimethylamino)phenyl]-N-hydroxy-4,6-dimethyl-7-oxohepta-2,4-dienamide Molecular Formula: C17H22N2O3 Molecular Weight: 302.36818 Category: Hydroxamic acid Mechanism: Class I HDAC inhibitor (HDAC1, 2, 3); Class IIa HDAC inhibitor (HDAC4, 7, 9); Class IIb inhibitor (HDAC6) Effect: Antifungal agent; Antibacterial agent; Histone deacetylase inhibitor; Protein synthesis inhibitor; Antineoplastic; Memory improvement; Rescue of CA3-CA1 LTP in APP/PS1 transgenic models |
Pathogenic genes: BCL2 Mechanistic genes: BCL2, HDACs, IL8, IL12A,IL12B, NFKB2, RARB Metabolic genes: Substrate: CYP3A4 (mayor) Inhibitor: HDACs Inducer: CYP1A1, CYP1B1, CYP2B6, CYP2E1, CYP7A1, SLC19A3 Transporter genes: SLC19A3 |
 |
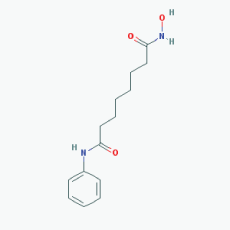
Name:Valproic Acid, 2-Propylpentanoic acid, Depakene, Depakine, Ergenyl, Dipropylacetic acid, Mylproin, Convulex, Myproic Acid IUPAC Name: 2-Propylpentanoic acid Molecular Formula: C8H16O2 Molecular Weight: 144.21144 Category: Short-chain fatty acid Mechanism: Class I HDAC inhibitor (HDAC1, 2, 3, 8) Effect: Anticonvulsant; Mood stabilizer; Antimanic agent; Enzyme inhibitor; Histone deacetylase inhibitor; GABA modulator; Memory improvement; Aβ and pTau decrease; CDK5 inactivation |
Pathogenic genes: CREB1, IL6, LEP, SCN2A, TGFB1, TNF, TRNK Mechanistic genes: ABAT, CDK5, GSK3B, HDAC1, HDAC2, HDAC3, HDAC8, HDAC9, LEP, LEPR, SCNs, SMN2 Metabolic genes: Substrate: ABCB1, CYP1A1 (minor), CYP2A6 (major), CYP2B6 (minor), CYP2C9 (major), CYP2C19 (minor), CYP2E1 (minor), CYP3A4 (minor), CYP4B1 (major), CYP4F2 (minor), UGT1A4, UGT1A6, UGT1A8, UGT1A9, UGT1A10, UGT2B7 Inhibitor: ABCB1, ACADSB, AKR1A1, CYP2A6 (moderate), CYP2C9 (strong), CYP2C19 (moderate), CYP2D6 (weak), CYP3A4 (moderate), HDAC1, HDAC2, HDAC3, HDAC8, HDAC9, UGT1A9, UGT2B1, UGT2B7 Inducer: ABCB1, AKR1C4, CASR, CYP2A6, CYP2B6, CYP3A4, CYP7A1, MAOA, NR1I2, SLC5A5, SLC6A2, SLC12A3, SLC22A16 Transporter genes: ABCB1, ABCC2, ABCG1, ABCG2, SCNs, SLC5A5, SLC6A2, SLC12A3, SLC22A16 Pleiotropic genes: ABL2, AGPAT2, ASL, ASS1, CDK4, CHRNA1, COL1A1, CPS1, CPT1A, DRD4, FMR1, FOS, HBB, HFE, HLA-A, HLA-B, ICAM1, IFNG, IL6, IL10, LEPR, NAGS, NR3C1, OTC, PTGES, STAT3, TGFB1, TNF, TP53. |
 |
Name: Vorinostat, Suberoylanilide hydroxamic acid(SAHA), Zolinza, Suberanilohydroxamic acid, 149647-78-9, N-hydroxy-N'-phenyloctanediamide, SAHA cpd IUPAC Name: N'-Hydroxy-N-phenyloctanediamide Molecular Formula: C14H20N2O3 Molecular Weight: 264.3202 Category: Hydroxamic acid Mechanism: Class I HDAC inhibitor (HDAC1, 2, 3, 8) Class IIb inhibitor (HDAC6) Effect: Antineoplastic, Memory improvement |
Pathogenic genes: BIRC3, CCND1, CDKN1A, CFLAR, CYP19A1, ERBB2, ERBB3, EGFR, RB1, TP53, TNF Mechanistic genes: CDKN1A, EGFR, ERBB2, ERBB3, STATs, TYMS, VEGFs Metabolic genes: Substrate: CYP2A6 (minor), CYP2C9 (minor), CYP2C19 (major), CYP2D6 (minor), CYP3A4 (major) Inhibitor: HDAC1, HDAC2, HDAC3, HDAC6 Inducer: CYP1A1, CYP1A2, CYP1B1 Pleiotropic genes: ALPs, TNF, TYMS |
ABAT: 4-aminobutyrate aminotransferase; ABCA1: ATP-binding cassette, sub-family A (ABC1), member 1; ABCB1: ATP-binding cassette, sub-family B (MDR/TAP), member 1; ABCC1: ATP-binding cassette, sub-family C (CFTR/MRP), member 1; ABCC2: ATP-binding cassette, sub-family C (CFTR/MRP), member 2; ABCC3: ATP-binding cassette, sub-family C (CFTR/MRP), member 3; ABCC4: ATP-binding cassette, sub-family C (CFTR/MRP), member 4; ABCC8: ATP-binding cassette, sub-family C (CFTR/MRP), member 8; ABCG1: ATP-binding cassette, sub-family G (WHITE), member 1; ABCG2: ATP-binding cassette, sub-family G (WHITE), member 2 (Junior blood group); ABCs: ATP-binding cassette family; ABL2: ABL proto-oncogene 2, non-receptor tyrosine kinase; ACADSB: acyl-CoA dehydrogenase, short/branched chain; AGPAT2: 1-acylglycerol-3-phosphate O-acyltransferase 2; AKR1A1: aldo-keto reductase family 1, member A1 (aldehyde reductase); AKR1C4: aldo-keto reductase family 1, member C4; AKT1: v-akt murine thymoma viral oncogene homolog 1; ALDH1A1: aldehyde dehydrogenase 1 family, member A1; ALDH3A1: aldehyde dehydrogenase 3 family, member A1; ALPs: alkaline phosphataseS; AMD1: adenosylmethionine decarboxylase 1; APAF1: apoptotic peptidase activating factor 1; APP: amyloid beta (A4) precursor protein; ARG1: arginase 1; ASL: argininosuccinate lyase; ASS1: argininosuccinate synthase 1; ATF3: activating transcription factor 3; BACE1: beta-site APP-cleaving enzyme 1; BAK1: BCL2-antagonist/killer 1; BAX: BCL2-associated X protein; BBC3: BCL2 binding component 3; BCL2: B-cell CLL/lymphoma 2; BCL2L1: BCL2-like 1; BCL2L11: BCL2-like 11 (apoptosis facilitator); BCR-ABL: BCR-ABL tyrosine kinase fusion; BDNF: brain-derived neurotrophic factor; BIRC3: baculoviral IAP repeat containing 3; BIRC5: baculoviral IAP repeat containing 5; BLK:BLK proto-oncogene, Src family tyrosine kinase; BRCA1: breast cancer 1, early onset; CASP3: caspase 3, apoptosis-related cysteine peptidase; CASR: calcium-sensing receptor; CAT: catalase; CAV1: caveolin 1, caveolae protein, 22kDa; CBS: cystathionine-beta-synthase; CCDN1: cyclin D1; CDA : cytidine deaminase; CDH1: cadherin 1, type 1; CDK4: cyclin-dependent kinase 4; CDK5: cyclin-dependent kinase 5; CDKN1A: cyclin-dependent kinase inhibitor 1A (p21, Cip1); CDKN2A: cyclin-dependent kinase inhibitor 2A; CDKN2B: cyclin-dependent kinase inhibitor 2B (p15, inhibits CDK4); CDKs: cyclin-dependent kinases; CFLAR: CASP8 and FADD-like apoptosis regulator; CFTR: cystic fibrosis transmembrane conductance regulator (ATP-binding cassette sub-family C, member 7); CHRNA1: cholinergic receptor, nicotinic, alpha 1 (muscle); COL1A1: collagen, type I, alpha 1; COMT: catechol-O-methyltransferase; CPS1: carbamoyl-phosphate synthase 1, mitochondrial; CPT1A: carnitine palmitoyltransferase 1A (liver); CREB1: cAMP responsive element binding protein 1; CTNNB1: catenin (cadherin-associated protein), beta 1, 88kDa; CYP19A1: cytochrome P450, family 19, subfamily A, polypeptide 1; CYP1A1: cytochrome P450, family 1, subfamily A, polypeptide 1; CYP1A2: cytochrome P450, family 1, subfamily A, polypeptide 2; CYP1B1: cytochrome P450, family 1, subfamily B, polypeptide 1; CYP2A6: cytochrome P450, family 2, subfamily A, polypeptide 6; CYP2C19: cytochrome P450, family 2, subfamily C, polypeptide 19; CYP2C8: cytochrome P450, family 2, subfamily C, polypeptide 8; CYP2C9: cytochrome P450, family 2, subfamily C, polypeptide 9; CYP2D6: cytochrome P450, family 2, subfamily D, polypeptide 6; CYP2E1: cytochrome P450, family 2, subfamily E, polypeptide 1; CYP3A4: cytochrome P450, family 3, subfamily A, polypeptide 4; CYP3A5: cytochrome P450, family 3, subfamily A, polypeptide 5; CYP4B1: cytochrome P450, family 4, subfamily B, polypeptide 1; CYP4F2: cytochrome P450, family 4, subfamily F, polypeptide 2; CYP7A1: cytochrome P450, family 7, subfamily A, polypeptide 1; DAPK1: death-associated protein kinase 1; DCK: deoxycytidine kinase; DNMT1: DNA (cytosine-5-)-methyltransferase 1; DNMT3A: DNA (cytosine-5-)-methyltransferase 3 alpha; DNMT3B: DNA (cytosine-5-)-methyltransferase 3 beta; DPYD: dihydropyrimidine dehydrogenase; DRD4: dopamine receptor D4; EDN1: endothelin 1; EGF: epidermal growth factor; EGFR: epidermal growth factor receptor; EP300: E1A binding protein p300; ERBB2: erb-b2 receptor tyrosine kinase 2; ERBB3: erb-b2 receptor tyrosine kinase 3; ERK:elk-related tyrosine kinase; ESR1: estrogen receptor 1; ESR2: estrogen receptor 2 (ER beta); FLT1: fms-related tyrosine kinase 1; FMR1: fragile X mental retardation 1; FOS: FBJ osteosarcoma oncogene; FSHR: follicle stimulating hormone receptor; GCLC: glutamate-cysteine ligase, catalytic subunit; GNMT: glycine N-methyltransferase; GRIN1: glutamate receptor, ionotropic, N-methyl D-aspartate 1; GRIN2B: glutamate receptor, ionotropic, N-methyl D-aspartate 2B; GSK3B: glycogen synthase kinase 3 beta; GSS: glutathione synthetase; GSTP1: glutathione S-transferase pi 1; HBB: hemoglobin, beta; HBG1: hemoglobin, gamma A; HDAC1: histone deacetylase 1; HDAC11: histone deacetylase 11; HDAC2: histone deacetylase 2; HDAC3: histone deacetylase 3; HDAC4: histone deacetylase 4; HDAC6: histone deacetylase 6; HDAC8: histone deacetylase 8; HDAC9: histone deacetylase 9; HDACs: histone deacetylases; HFE: hemochromatosis; HIF1A: hypoxia inducible factor 1, alpha subunit (basic helix-loop-helix transcription factor); HIST3H3: histone cluster 3, H3; HIST4H4: histone cluster 4, H4; HLA-A : major histocompatibility complex, class I, A; HLA-B: major histocompatibility complex, class I, B; HSP90As: heat shock protein 90kDa alpha (cytosolic), class A; HSPA8: heat shock 70kDa protein 8; HTR3A: 5-hydroxytryptamine (serotonin) receptor 3A, ionotropic; ICAM1: intercellular adhesion molecule 1; IFNG: interferon, gamma; IL10: interleukin 10; IL12: interleukin 12; IL12A: interleukin 12A; IL12B: interleukin 12B; IL1A: interleukin 1, alpha; IL23A: interleukin 23, alpha subunit p19; IL6: interleukin 6; IL8: interleukin 8; JUN: jun proto-oncogene; KDR: kinase insert domain receptor; KLRK1: killer cell lectin-like receptor subfamily K, member 1; LEP: leptin; LEPR: leptin receptor; MAOA: monoamine oxidase A; MAT1A: methionine adenosyltransferase I, alpha; MGMT: O-6-methylguanine-DNA methyltransferase; MLH1: mutL homolog 1; MMP2: matrix metallopeptidase 2; MMP9: matrix metallopeptidase 9; MMPs: matrix metallopeptidases; MSH2: mutS homolog 2; MSR1: macrophage scavenger receptor 1; MYC: v-myc avian myelocytomatosis viral oncogene homolog; NAGS: N-acetylglutamate synthase; NF2:neurofibromin 2 (merlin); NFKB1:nuclear factor of kappa light polypeptide gene enhancer in B-cells 1; NFKB2:nuclear factor of kappa light polypeptide gene enhancer in B-cells 2 (p49/p100); NOS2: nitric oxide synthase 2, inducible; NOS3:nitric oxide synthase 3 (endothelial cell); NQO1:NAD(P)H dehydrogenase, quinone 1; NQO2:NAD(P)H dehydrogenase, quinone 2; NR1I2:nuclear receptor subfamily 1, group I, member 2; NR1I3:nuclear receptor subfamily 1, group I, member 3; NR3C1: nuclear receptor subfamily 3, group C, member 1 (glucocorticoid receptor); NT3:3'-nucleotidase; NTRK2:neurotrophic tyrosine kinase, receptor, type 2; OTC:ornithine carbamoyltransferase; P2RY2:purinergic receptor P2Y, G-protein coupled, 2; PDGFRB:platelet-derived growth factor receptor, beta polypeptide; PDGFRs: platelet-derived growth factor receptors; PLA2R1:phospholipase A2 receptor 1, 180kDa; PLCB1: phospholipase C, beta 1 (phosphoinositide-specific); PMAIP1: phorbol-12-myristate-13-acetate-induced protein 1;PRKAs: protein kinase family, AMP-activated; PSEN1:presenilin 1; PTGES: prostaglandin E synthase; PTGS1: prostaglandin-endoperoxide synthase 1 (prostaglandin G/H synthase and cyclooxygenase); PTGS2:prostaglandin-endoperoxide synthase 2 (prostaglandin G/H synthase and cyclooxygenase); RARB:retinoic acid receptor, beta; RASSF1:Ras association (RalGDS/AF-6) domain family member 1; RB1: retinoblastoma 1; RRM1: ribonucleotide reductase M1; ROS1: ROS proto-oncogene 1 , receptor tyrosine kinase; RRM1: ribonucleotide reductase M1; RRM2:ribonucleotide reductase M2; RYR1:ryanodine receptor 1 (skeletal); SCN2A: sodium channel, voltage gated, type II alpha subunit; SCNs: sodium channel family; SIRT1: sirtuin 1; SIRT2: sirtuin 2; SIRT3:sirtuin 3; SIRT5:sirtuin 5; SLC12A3: solute carrier family 12 (sodium/chloride transporter), member 3; SLC15s: solute carrier family 15; SLC19A3:solute carrier family 19 (thiamine transporter), member 3; SLC19A3:solute carrier family 19 (thiamine transporter), member 3; SLC22A16:solute carrier family 22 (organic cation/carnitine transporter), member 16; SLC22s: solute carrier family 22; SLC25A26: solute carrier family 25 (S-adenosylmethionine carrier), member 26; SLC28A1:solute carrier family 28 (concentrative nucleoside transporter), member 1; SLC29A1: solute carrier family 29 (equilibrative nucleoside transporter), member 1; SLC29As: solute carrier family 29; SLC5A5:solute carrier family 5 (sodium/iodide cotransporter), member 5; SLC6A2:solute carrier family 6 (neurotransmitter transporter), member 2; SLCO1B3:solute carrier organic anion transporter family, member 1B3; SMN2:survival of motor neuron 2, centromeric; SNCA: synuclein, alpha (non A4 component of amyloid precursor; SOCS1: suppressor of cytokine signaling 1; SOCS3:suppressor of cytokine signaling 3; SRC:SRCproto-oncogene, non-receptor tyrosine kinase; SREBF1:sterol regulatory element binding transcription factor 1; SRM:spermidine synthase; STAT1:signal transducer and activator of transcription 1, 91kDa; STAT3: signal transducer and activator of transcription 3 (acute-phase response factor); STATs: signal transducer and activator of transcription family; SULT1C2:sulfotransferase family, cytosolic, 1C, member 2; TGFB1: transforming growth factor, beta 1; TIMP3:TIMP metallopeptidase inhibitor 3; TLR3:toll-like receptor 3; TNF:tumor necrosis factor; TNFRSF10A: tumor necrosis factor receptor superfamily, member 10a; TNFRSF10B:tumor necrosis factor receptor superfamily, member 10b; TNFRSF1B:tumor necrosis factor receptor superfamily, member 1B; TNFSF10:tumor necrosis factor (ligand) superfamily, member 10; TP53: tumor protein p53; TPMT:thiopurine S-methyltransferase; TRNK: mitochondrially encoded tRNA lysine; TRPs: transient receptor potential cation channels; TYMS:thymidylate synthetase; UCK1:uridine-cytidine kinase 1; UCK2: uridine-cytidine kinase 2; UGT1A10:UDP glucuronosyltransferase 1 family, polypeptide A10; UGT1A4:UDP glucuronosyltransferase 1 family, polypeptide A4; UGT1A6:UDP glucuronosyltransferase 1 family, polypeptide A6; UGT1A8:UDP glucuronosyltransferase 1 family, polypeptide A8; UGT1A9:UDP glucuronosyltransferase 1 family, polypeptide A9; UGT2B1: UDP glucuronosyltransferase 1 family, polypeptide B1; UGT2B7:UDP glucuronosyltransferase 2 family, polypeptide B7; VCAM1:vascular cell adhesion molecule 1; VEGFA: vascular endothelial growth factor A; VEGFs: vascular endothelial growth factor family; VHL:von Hippel-Lindau tumor suppressor, E3 ubiquitin protein ligase.
Source: Adapted from Cacabelos (Ref. 17) and Cacabelos and Torrellas (Ref. 15).
Table 3: Pharmacological profile and pharmacogenetics of selected epigenetic drugs.
| Drug | Properties | Pharmacogenetics |
|---|---|---|
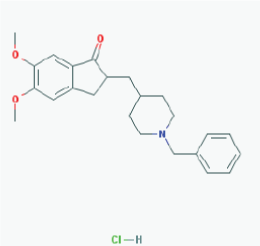 |
Name: Donepezil hydrochloride, Aricept, 120011-70-3, Donepezil HCl, BNAG, E-2020, E2020 IUPAC Name: 2-[(1-benzylpiperidin-4-yl)methyl]-5,6-dimethoxy-2,3-dihydroinden-1-one;hydrochloride Molecular Formula: C24H30ClNO3 Molecular Weight: 415.9529 g/mol Category: Cholinesterase inhibitor Mechanism: Centrally active, reversible acetylcholinesterase inhibitor; increases the acetylcholine available for synaptic transmission in the CNS Effect: Nootropic agent, cholinesterase inhibitor, parasympathomimetic effect |
Pathogenic genes: APOE, CHAT Mechanistic genes: CHAT, ACHE, BCHE Drug metabolism-related genes: - Substrate: CYP2D6 (major), CYP3A4 (major), UGTsACHE - Inhibitor: ACHE, BCHE Transporter genes: ABCB1 |
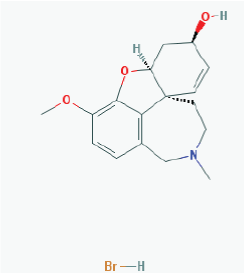 |
Name: Galantamine hydrobromide, Galanthamine hydrobromide, 1953-04-4, Nivalin, Razadyne, UNII-MJ4PTD2VVW, Nivaline IUPAC Name: (1S,12S,14R)-9-methoxy-4-methyl-11-oxa-4-azatetracyclo[8.6.1.0^{1,12}.0^{6,17}]heptadeca-6,8,10(17),15-tetraen-14-ol Molecular Formula: C17H22BrNO3 Molecular Weight: 368.26548 g/mol Category: Cholinesterase inhibitor Mechanism: Reversible and competitive acetylcholinesterase inhibition leading to an increased concentration of acetylcholine at cholinergic synapses; modulates nicotinic acetylcholine receptor; may increase glutamate and serotonin levels Effect: Nootropic agent, cholinesterase inhibitor, parasympathomimetic effect |
Pathogenic genes: APOE, APP Mechanisticgenes: ACHE, BCHE, CHRNA4, CHRNA7, CHRNB2 Drug metabolism-related genes: - Substrate: CYP2D6 (major), CYP3A4 (major), UGT1A1 - Inhibitor: ACHE, BCHE |
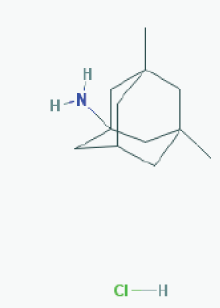 |
Name: Memantine Hydrochloride, 41100-52-1, Namenda, Memantine HCL, Axura, 3,5-Dimethyl-1-adamantanamine hydrochloride, 3,5-dimethyladamantan-1-amine hydrochloride IUPAC Name: 3,5-dimethyladamantan-1-amine;hydrochloride Molecular Formula: C12H22ClN Molecular Weight: 215.76278 g/mol Category: N-Methyl-D-Aspartate receptor antagonist Mechanism: Binds preferentially to NMDA receptor-operated cation channels; may act by blocking actions of glutamate, mediated in part by NMDA receptors Effect: Dopamine agent, antiparkinson agent, excitatory amino acid antagonist, antidyskinetic |
Pathogenic genes: APOE, MAPT, PSEN1 Mechanistic genes: CHRFAM7A, DLGAP1, FOS, GRIN2A, GRIN2B, GRIN3A, HOMER1, HTR3A Drug metabolism-related genes: -Inhibitor: CYP1A2 (weak), CYP2A6 (weak), CYP2B6 (strong), CYP2C9 (weak), CYP2C19 (weak), CYP2D6 (strong), CYP2E1 (weak), CYP3A4 (weak), NR1I2 Transporter genes: NR1I2 Pleiotropic genes: APOE, MAPT, MT-TK, PSEN1 |
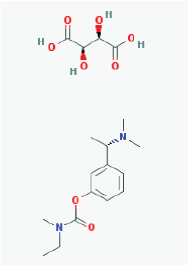 |
Name: Rivastigmine tartrate, 129101-54-8, SDZ-ENA 713, Rivastigmine hydrogentartrate, Rivastigmine Hydrogen Tartrate, ENA 713, ENA-713 IUPAC Name: (2R,3R)-2,3-dihydroxybutanedioic acid;[3-[(1S)-1-(dimethylamino)ethyl]phenyl] N-ethyl-N-methylcarbamate Molecular Formula: C18H28N2O8 Molecular Weight: 400.42352 g/mol Category: Cholinesterase inhibitor Mechanism: Increases acetylcholine in CNS through reversible inhibition of its hydrolysis by cholinesterase Effect: Neuroprotective agent, cholinesterase inhibitor, cholinergic agent |
Pathogenic genes: APOE, APP, CHAT Mechanistic genes: ACHE, BCHE, CHAT, CHRNA4, CHRNB2 Drug metabolism-related genes: -Inhibitor: ACHE, BCHE Pleiotropic genes: APOE, MAPT |
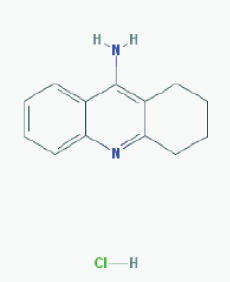 |
Name:Tacrine Hydrochloride, Tacrine HCl, 1684-40-8, Hydroaminacrine, tacrine.HCl, 9-AMINO-1,2,3,4-TETRAHYDROACRIDINE HYDROCHLORIDE, Tenakrin IUPAC Name: 1,2,3,4-tetrahydroacridin-9-amine;hydrochloride Molecular Formula: C13H15ClN2 Molecular Weight: 234.7246 g/mol Category: Cholinesterase inhibitor Mechanism: Elevates acetylcholine in cerebral cortex by slowing degradation of acetylcholine Effect: Nootropic agent, cholinesterase inhibitor, Parasympathomimetic effect |
Pathogenic genes: APOE Mechanistic genes: ACHE, BCHE, CHRNA4, CHRNB2 Drug metabolism-related genes: -Substrate: CYP1A2 (major), CYP2D6 (minor), CYP3A4 (major) -Inhibitor: ACHE, BCHE, CYP1A2 (weak) Transporter genes: SCN1A Pleiotropic genes: APOE, CES1, GSTM1, GSTT1, LEPR, MTHFR |
ADH1A: Alcohol dehydrogenase 1A (class I), alpha polypeptide; AADAC: Arylacetamide deacetylase; AANAT: aralkylamine N-acetyltransferase; ACSL1: Acyl-CoA synthetase long-chain family member 1; ACSL3: Acyl-CoA synthetase long-chain family member 3; ACSL4: Acyl-CoA synthetase long-chain family member 4; ACSM1: Acyl-CoA synthetase medium-chain family member 1; ACSM2B: Acyl-CoA synthetase medium-chain family member 2B; ACSM3: Acyl-CoA synthetase medium-chain family, member 3; ADH1B: Alcohol dehydrogenase 1B (class I), beta polypeptide; ADH1C: Alcohol dehydrogenase 1C (class I), gamma polypeptide; ADH4: Alcohol dehydrogenase 4 (class II), pi polypeptide; ADH5: Alcohol dehydrogenase 5 (class III), chi polypeptide; ADH6: Alcohol dehydrogenase 6 (class V); ADH7: Alcohol dehydrogenase 7 (class IV), mu or sigma polypeptide; ADHFE1: Alcohol dehydrogenase, iron containing, 1; AGXT: Alanine-glyoxylate aminotransferase; AKR1A1: Aldo-keto reductase family 1, member A1 (aldehyde reductase); AKR1B1: Aldo-keto reductase family 1, member B1 (aldose reductase); AKR1C1: Aldo-keto reductase family 1, member C1; AKR1D1: Aldo-keto reductase family 1, member D1; ALDH1A1: Aldehyde dehydrogenase 1 family, member A1; ALDH1A2: Aldehyde dehydrogenase family 1, subfamily A2; ALDH1A3: Aldehyde dehydrogenase family 1, subfamily A3; ALDH1B1: Aldehyde dehydrogenase 1 family, member B1; ALDH2: Aldehyde dehydrogenase 2 family (mitochondrial); ALDH3A1: Aldehyde dehydrogenase 3 family, member A1; ALDH3A2: Aldehyde dehydrogenase 3 family, member A2; ALDH3B1: Aldehyde dehydrogenase 3 family, member B1; ALDH3B2: Aldehyde dehydrogenase 3 family, member B2; ALDH4A1: Aldehyde dehydrogenase 4 family, member A1; ALDH5A1: Aldehyde dehydrogenase 5 family, member A1; ALDH6A1: Aldehyde dehydrogenase 6 family, member A1; ALDH7A1: Aldehyde dehydrogenase 7 family, member A1; ALDH8A1: Aldehyde dehydrogenase 8 family, member A1; ALDH9A1: Aldehyde dehydrogenase 9 family, member A1; AOX1: Aldehyde oxidase 1; AS3MT: Arsenic (+3 oxidation state) methyltransferase; ASMT: Acetylserotonin O-methyltransferase; BAAT: Bile acid CoA: amino acid N-acyltransferase (glycine N-choloyltransferase); CBR1: Carbonyl reductase 1; CBR3: Carbonyl reductase 3;CBR4: Carbonyl reductase 4; CCBL1: Cysteine conjugate-beta lyase, cytoplasmic; CDA: Cytidine deaminase; CEL: Carboxyl ester lipase; CES1: Carboxylesterase 1; CES1P1: Carboxylesterase 1 pseudogene 1; CES2: Carboxylesterase 2; CES3: Carboxylesterase 3; CES5A: Carboxylesterase 5A; CHST1: Carbohydrate (keratan sulfate Gal-6) sulfotransferase 1; CHST2: Carbohydrate (N-acetylglucosamine-6-O) sulfotransferase 2; CHST3: Carbohydrate (chondroitin 6) sulfotransferase 3; CHST4: Carbohydrate (N-acetylglucosamine 6-O) sulfotransferase 4; CHST5: Carbohydrate (N-acetylglucosamine 6-O) sulfotransferase 5; CHST6: Carbohydrate (N-acetylglucosamine 6-O) sulfotransferase 6; CHST7: Carbohydrate (N-acetylglucosamine 6-O) sulfotransferase 7; CHST8: Carbohydrate (N-acetylgalactosamine 4-0) sulfotransferase 8; CHST9: Carbohydrate (N-acetylgalactosamine 4-0) sulfotransferase 9; CHST10: Carbohydrate sulfotransferase 10; CHST11: Carbohydrate (chondroitin 4) sulfotransferase 11; CHST12: Carbohydrate (chondroitin 4) sulfotransferase 12; CHST13: Carbohydrate (chondroitin 4) sulfotransferase 13; COMT: Catechol-O-methyltransferase; CYB5R3: Cytochrome b5 reductase 3; CYP1A1: Cytochrome P450, family 1, subfamily A, polypeptide 1; CYP1A2: Cytochrome P450, family 1, subfamily A, polypeptide 2; CYP1B1: Cytochrome P450, family 1, subfamily B, polypeptide 1; CYP2A6: Cytochrome P450, family 2, subfamily A, polypeptide 6; CYP2A7: Cytochrome P450, family 2, subfamily A, polypeptide 7; CYP2A13: Cytochrome P450, family 2, subfamily A, polypeptide 13; CYP2B6: Cytochrome P450, family 2, subfamily B, polypeptide 6; CYP2C8: Cytochrome P450, family 2, subfamily C, polypeptide 8; CYP2C9: Cytochrome P450, family 2, subfamily C, polypeptide 9; CYP2C18: Cytochrome P450, family 2, subfamily C, polypeptide 18; CYP2C19: Cytochrome P450, family 2, subfamily C, polypeptide 19; CYP2D6: Cytochrome P450, family 2, subfamily D, polypeptide 6; CYP2D7P1: Cytochrome P450, family 2, subfamily D, polypeptide 7 pseudogene 1; CYP2E1: Cytochrome P450, family 2, subfamily E, polypeptide 1; CYP2F1: Cytochrome P450, family 2, subfamily F, polypeptide 1; CYP2J2: Cytochrome P450, family 2, subfamily J, polypeptide 2; CYP2R1: Cytochrome P450, family 2, subfamily R, polypeptide 1; CYP2S1: Cytochrome P450, family 2, subfamily S, polypeptide 1; CYP2W1: Cytochrome P450, family 2, subfamily W, polypeptide 1; CYP3A4: Cytochrome P450, family 3, subfamily A, polypeptide 4; CYP3A5: Cytochrome P450, family 3, subfamily A, polypeptide 5; CYP3A7: Cytochrome P450, family 3, subfamily A, polypeptide 7; CYP3A43: Cytochrome P450, family 3, subfamily A, polypeptide 43; CYP4A11: Cytochrome P450, family 4, subfamily A, polypeptide 11; CYP4A22: Cytochrome P450, family 4, subfamily A, polypeptide 22; CYP4B1: Cytochrome P450, family 4, subfamily B, polypeptide 1; CYP4F2: Cytochrome P450, family 4, subfamily F, polypeptide 2; CYP4F3: Cytochrome P450, family 4, subfamily F, polypeptide 3; CYP4F8: Cytochrome P450, family 4, subfamily F, polypeptide 8; CYP4F11: Cytochrome P450, family 4, subfamily F, polypeptide 11; CYP4F12: Cytochrome P450, family 4, subfamily F, polypeptide 12; CYP4Z1: Cytochrome P450, family 4, subfamily Z, polypeptide 1; CYP7A1: Cytochrome P450, family 7, subfamily A, polypeptide 1; CYP7B1: Cytochrome P450, family 7, subfamily B, polypeptide 1; CYP8B1: Cytochrome P450, family 8, subfamily B, polypeptide 1; CYP11A1: Cytochrome P450, family 11, subfamily A, polypeptide 1; CYP11B1: Cytochrome P450, family 11, subfamily B, polypeptide 1: CYP11B2: Cytochrome P450, family 11, subfamily B, polypeptide 2; CYP17A1: Cytochrome P450, family 17, subfamily A, polypeptide 1; CYP19A1: Cytochrome P450, family 19, subfamily A, polypeptide 1; CYP20A1: Cytochrome P450, family 20, subfamily A, polypeptide 1; CYP21A2: Cytochrome P450, family 21, subfamily A, polypeptide 2; CYP24A1: Cytochrome P450, family 24, subfamily A, polypeptide 1; CYP26A1: Cytochrome P450, family 26, subfamily A, polypeptide 1; CYP26B1: Cytochrome P450, family 26, subfamily B, polypeptide 1; CYP26C1: Cytochrome P450, family 26, subfamily C, polypeptide 1; CYP27A1: Cytochrome P450, family 27, subfamily A, polypeptide 1; CYP27B1: Cytochrome P450, family 27, subfamily B, polypeptide 1; CYP39A1: Cytochrome P450, family 39, subfamily A, polypeptide 1; CYP46A1: Cytochrome P450, family 46, subfamily A, polypeptide 1; CYP51A1: Cytochrome P450, family 51, subfamily A, polypeptide 1; DDOST: Dolichyl-diphosphooligosaccharide--protein glycosyltransferase subunit (non-catalytic); DHRS1: Dehydrogenase/reductase (SDR family) member 1; DHRS2: Dehydrogenase/reductase (SDR family) member 2; DHRS3: Dehydrogenase/reductase (SDR family) member 3; DHRS4: Dehydrogenase/reductase (SDR family) member 4; DHRS7: Dehydrogenase/reductase (SDR family) member 7; DHRS9: Dehydrogenase/reductase (SDR family) member 9; DHRS12: Dehydrogenase/reductase (SDR family) member 12; DHRS13: Dehydrogenase/reductase (SDR family) member 13; DHRSX: Dehydrogenase/reductase (SDR family) X-linked; DLGAP1: discs, large (Drosophila) homolog-associated protein 1; DPEP1: Dipeptidase 1 (renal); DPYD: Dihydropyrimidine dehydrogenase; EPHX1: Epoxide hydrolase 1, microsomal (xenobiotic); EPHX2: Epoxide hydrolase 2, microsomal (xenobiotic); ESD: Esterase D; FMO1: Flavin containing monooxygenase 1; FMO2: Flavin containing monooxygenase 2; FMO3: Flavin containing monooxygenase 3; FMO4: Flavin containing monooxygenase 4; FMO5: Flavin containing monooxygenase 5; FMO6P: Flavin containing monooxygenase 6 pseudogene; FOS: FBJ murine osteosarcoma viral oncogene homolog; GAL3ST1: Galactose-3-O-sulfotransferase 1; GAMT: Guanidinoacetate N-methyltransferase; GLRX: Glutaredoxin (thioltransferase); GLYAT: Glycine-N-acyltransferase; GNMT: Glycine N-methyltransferase; GPX1: Glutathione peroxidase 1; GPX2: Glutathione peroxidase 2 (gastrointestinal); GPX3: Glutathione peroxidase 3 (plasma); GPX4: Glutathione peroxidase 4; GPX5: Glutathione peroxidase 5; GPX6: Glutathione peroxidase 6 (olfactory); GPX7: Glutathione peroxidase 7; GSR: Glutathione reductase; GSTA1: Glutathione S-transferase alpha 1; GSTA2: Glutathione S-transferase alpha 2; GSTA3: Glutathione S-transferase alpha 3; GSTA4: Glutathione S-transferase alpha 4; GSTA5: Glutathione S-transferase alpha 5; GSTCD: Glutathione S-transferase, C-terminal domain containing; GSTK1: Glutathione S-transferase kappa 1; GSTM1: Glutathione S-transferase mu 1; GSTM2: Glutathione S-transferase mu 2 (muscle); GSTM3: Glutathione S-transferase mu 3 (brain); GSTM4: Glutathione S-transferase mu 4; GSTM5: Glutathione S-transferase mu 5; GSTO1: Glutathione S-transferase omega 1; GSTO2: Glutathione S-transferase omega 2; GSTP1: Glutathione S-transferase pi 1; GSTT1: Glutathione S-transferase theta 1; GSTT2: Glutathione S-transferase theta 2; GSTZ1: Glutathione S-transferase zeta 1; GZMA: Granzyme A (granzyme 1, cytotoxic T-lymphocyte-associated serine esterase 3; GZMB: Granzyme B (granzyme 2, cytotoxic T-lymphocyte-associated serine esterase 1); HNMT: Histamine N-methyltransferase; HOMER1: homer homolog 1 (Drosophila);HSD11B1: Hydroxysteroid (11-beta) dehydrogenase 1; HSD17B10: Hydroxysteroid (17-beta) dehydrogenase 10; HSD17B11: Hydroxysteroid (17-beta) dehydrogenase 11; HSD17B14: Hydroxysteroid (17-beta) dehydrogenase 14; INMT: Indolethylamine N-methyltransferase; MAOA: Monoamine oxidase A; MAOB: monoamine oxidase B; METAP1: Methionyl aminopeptidase 1; MGST1: Microsomal glutathione S-transferase 1; MGST2: Microsomal glutathione S-transferase 1; MGST3: Microsomal glutathione S-transferase 3; NAA20: N(alpha)-acetyltransferase 20, NatB catalytic subunit; NAT1: N-acetyltransferase 1 (arylamine N-acetyltransferase); NAT2: N-acetyltransferase 2 (arylamine N-acetyltransferase); NNMT: Nicotinamide N-methyltransferase; NQO1: NAD(P)H dehydrogenase, quinone 1; NQO2: NAD(P)H dehydrogenase, quinone 2; NR1I2: nuclear receptor subfamily 1, group I, member 2;PNMT: Phenylethanolamine N-methyltransferase; PON1: Paraoxonase 1; PON2: Paraoxonase 2; PON3: Paraoxonase 3; POR: P450 (cytochrome) oxidoreductase; PTGES: Prostaglandin E synthase; PTGS1: Prostaglandin-endoperoxide synthase 1 (prostaglandin G/H synthase and cyclooxygenase); PTGS2: Prostaglandin-endoperoxide synthase 2 (prostaglandin G/H synthase and cyclooxygenase); SAT1: Spermidine/spermine N1-acetyltransferase 1; SMOX: Spermine oxidase; SOD1: Superoxide dismutase 1, soluble; SOD2: Superoxide dismutase 2, mitochondrial; SULT1A1: Sulfotransferase family, cytosolic, 1A, phenol-preferring, member 1; SULT1A2: Sulfotransferase family, cytosolic, 1A, phenol-preferring, member 2; SULT1A3: Sulfotransferase family, cytosolic, 1A, phenol-preferring, member 3; SULT1B1: Sulfotransferase family, cytosolic, 1B, member 1; SULT1C1: Sulfotransferase family, cytosolic, 1C, member 1; SULT1C2: Sulfotransferase family, cytosolic, 1C, member 2; SULT1C3: Sulfotransferase family, cytosolic, 1C, member 3; SULT1C4: Sulfotransferase family, cytosolic, 1C, member 4; SULT1E1: Sulfotransferase family 1E, estrogen-preferring, member 1; SULT2A1: Sulfotransferase family, cytosolic, 2A, dehydroepiandrosterone (DHEA)-preferring, member 1; SULT2B1: Sulfotransferase family, cytosolic, 2B, member 1; SULT4A1: Sulfotransferase family 4A, member 1; SULT6B1: sulfotransferase family, cytosolic, 6B, member 1; TBXAS1: Thromboxane A synthase 1 (platelet); TPMT: Thiopurine S-methyltransferase; TST: Thiopurine S-methyltransferase; UCHL1: Ubiquitin carboxyl-terminal esterase L1 (ubiquitin thiolesterase); UCHL3: Ubiquitin carboxyl-terminal esterase L3 (ubiquitin thiolesterase); UGT1A1: UDP glucuronosyltransferase 1 family, polypeptide A1; UGT1A3: UDP glucuronosyltransferase 1 family, polypeptide A3; UGT1A4: UDP glucuronosyltransferase 1 family, polypeptide A4; UGT1A5: UDP glucuronosyltransferase 1 family, polypeptide A5; UGT1A6: UDP glucuronosyltransferase 1 family, polypeptide A6; UGT1A7: UDP glucuronosyltransferase 1 family, polypeptide A7; UGT1A8: UDP glucuronosyltransferase 1 family, polypeptide A8; UGT1A9: UDP glucuronosyltransferase 1 family, polypeptide A9; UGT1A10: UDP glucuronosyltransferase 1 family, polypeptide A10; UGT2A1: UDP glucuronosyltransferase 2 family, polypeptide A1, complex locus; UGT2A3: UDP glucuronosyltransferase 2 family, polypeptide A3; UGT2B10: UDP glucuronosyltransferase 2 family, polypeptide B10; UGT2B11: UDP glucuronosyltransferase 2 family, polypeptide B11; UGT2B15: UDP glucuronosyltransferase 2 family, polypeptide B15; UGT2B17: UDP glucuronosyltransferase 2 family, polypeptide B17; UGT2B28: UDP glucuronosyltransferase 2 family, polypeptide B28; UGT2B4: UDP glucuronosyltransferase 2 family, polypeptide B4; UGT2B7: UDP glucuronosyltransferase 2 family, polypeptide B7; UGT3A1: UDP glycosyltransferase 3 family, polypeptide A1; UGT8: UDP glycosyltransferase 8; XDH: Xanthine dehydrogenase.
Table 4: Pharmacological properties and pharmacogenomics of conventional anti-dementia drugs
The structural classification of HDAC inhibitors differentiates several classes: (i) short-chain fatty acids (sodium butfyrate, sodium phenyl butyrate, valproic acid, pivaloyloxymethyl butyrate (AN-9, Pivanex))(selective inhibitors of class I HDACs); (ii) hydroxamic acids (suberoylanilide hydroxamic acid (SAHA, Vorinostat), oxamflatin, pyroxamide, trichostatin A (TSA), m-carboxycinnamic acid bis-hydroxamide (CBHA), derivatives of the marine sponge Psammaplysilla purpurea (NVP-LAQ824, NVP-LBH589), LBH-589 (Panobinostat), ITF2357 (Givinostat), PXD101 (Belinostat), CHR- 3996, CHR-2845, PCI-24781)(inhibitors of class I and II HDACs); (iii) cyclic peptides (depsipeptide FR901228, romidepsin; apicidin, cyclic hydroxamic acid-containing peptides (CHAPS), cyclic tetrapetides trapoxin A and B with the epoxyketone-containing amino acid (2S,9S)- 2-amino-8-0xo-9,10-epoxy-decanoyl (Aoe), chlamydocin, HC toxin, bacterial FK228)(class I HDAC inhibitors); (iv) benzamides (MS- 275 (Entinostat), CI-994, RGFP136, MGCD0103 (Mocetinostat)) (class I HDAC inhibitors; selective HDAC1 and HDAC3 inhibitors); (v) ketones (trifluoromethyl ketone); (vi) sirtuin inhibitors (Class III HDAC inhibitors)(nicotinamide/niacinamide, suramin); and (vii) miscellaneous compounds (MGCD-0103, natural bioproducts) [15- 17,101] (Tables 2 and 3).
miRNAs exert regulatory control over mRNA stability and translation and may contribute to local and activity-dependent posttranscriptional control of synapse-associated mRNAs. miRNAs are small non-coding RNA regulators of protein synthesis that are essential for normal brain development and function. Their profiles are significantly altered in AD. miR-9 and -181c are down-regulated by Aβ in hippocampal cultures. The Aβ precursor protein APP itself is a target of miRNA regulation. The 3’ untranslated regions (3’ UTRs) of TGFBI, TRIM2, SIRT1 and BTBD3 are repressed by miR-9 and -181c, either alone or in combination. miRNA are integral components of the APP regulatory framework and potential targets for future AD therapeutics. Cohen et al. found a developmentally and activityregulated miRNA (miR-485) that controls dendritic spine number and synapse formation in an activity-dependent homeostatic manner. Many plasticity-associated genes contain predicted miR-485 binding sites. The presynaptic protein SV2A is a target of miR-485. miR-485 negatively regulates dendritic spine density, postsynaptic density 95 (PSD-95) clustering, and surface expression of GluR2. miR-485 overexpression reduced spontaneous synaptic responses and transmitter release. miRNA-485 and the presynaptic protein SV2A regulate homeostatic plasticity and CNS development, and their dysfunction might have possible implications in AD.
RNA interference (RNAi) technology may potentially be able to control AD, inhibiting the protein expression of specific genes by activating a sequence-specific RNA degradation process [108]. Short interfering nucleic acid (siNA), siRNA, dsRNA, miRNA and short hairpin RNA (shRNA) are capable of mediating RNA interference (RNAi) against BACE, APP, PS-1 and PS-2 gene expression [9]. RNAi-based treatments represent a promising therapeutic strategy for AD and other complex disorders. miRNA mimics, analogs of miRNA precursors, and anti-miRNAs are being explored as candidate therapeutic interventions for AD. Overexpression of miR-124 and miR- 195 may reduce Aβ levels by targeting BACE1 [109,110].
Pharmacoepigenomics
Pharmacogenomics account for 30-90% variability in pharmacokinetics and pharmacodynamics; however, pharmacogenetics alone does not predict all phenotypic variations in drug response. Individual differences in drug response are associated with genetic and epigenetic variability and disease determinants [111,112]. The genes involved in the pharmacogenomic response to drugs fall into five major categories: (i) genes associated with disease pathogenesis; (ii) genes associated with the mechanism of action of drugs (enzymes, receptors, transmitters, messengers); (iii) genes associated with drug metabolism: (a) phase I reaction enzymes: alcohol dehydrogenases, aldehyde dehydrogenases, aldo-keto reductases, amine oxidases, carbonyl reductases, cytidine deaminase, cytochrome P450 enzyme family, cytochrome b5 reductase, dihydroprimidine dehydrogenase, esterases, epoxidases, flavin-containing monooxygenases, glutathione reductase/peroxidases, short-chain dehydrogenases/reductases, superoxide dismutases, and xanthine dehydrogenase; and (b): phase II reaction enzymes: amino acid transferases, dehydrogenases, esterases, glucuronosyl transferases, glutathione transferases, methyl transferases, N-acetyl transferases, thioltransferase, and sulfotransferases; (iv) genes associated with drug transporters: ABC genes, especially ABCB1 (ATPbinding cassette, subfamily B, member 1; P-glycoprotein-1, P-gp1; Multidrug Resistance 1, MDR1), ABCC1, ABCG2 (White1), genes of the solute carrier superfamily (SLC) and solute carrier organic (SLCO) transporter family, responsible for the transport of multiple endogenous and exogenous compounds, including folate (SLC19A1), urea (SLC14A1, SLC14A2), monoamines (SLC29A4, SLC22A3), aminoacids (SLC1A5, SLC3A1, SLC7A3, SLC7A9, SLC38A1, SLC38A4, SLC38A5, SLC38A7, SLC43A2, SLC45A1), nucleotides (SLC29A2, SLC29A3), fatty acids (SLC27A1-6), neurotransmitters (SLC6A2 (noradrenaline transporter), SLC6A3 (dopamine transporter), SLC6A4 (serotonin transporter, SERT), SLC6A5, SLC6A6, SLC6A9, SLC6A11, SLC6A12, SLC6A14, SLC6A15, SLC6A16, SLC6A17, SLC6A18, SLC6A19), glutamate (SLC1A6, SLC1A7), and others); and (v) pleiotropic genes involved in multifaceted cascades and metabolic reactions [3,113-117] (Tables 3 and 4).
Epigenetic regulation is responsible for the tissue-specific expression of genes involved in pharmacogenetic processes, and epigenetics plays a key role in the development of drug resistance. Epigenetic changes affect cytochrome P450 enzyme expression, major transporter function, and nuclear receptor interactions [114-117]. Although this is a still poorly explored field, epigenetic regulation of genes encoding drug-metabolizing enzymes (CYP1A1, 1A2, 1B1, 1A6, 2A13, 2B6, 2C8, 2C9, 2C18, 2C19, 2D6, 2E1, 2J2, 2F1, 2R1, 2S1, 2W1, 3A4, 3A5, 3A7, 3A43, UGT1, GSTP1), drug transporters (ABCB1/MDR1/P-gp, ABCC1/MRP1, ABCC11/MRP8, ABCG2/BCRP, SLC19A1, SLC22A8), and nuclear receptors (RARB2, ESR1, NR1I2, HNF41) has been documented in pioneering studies of pharmacoepigenetics [111-118] (Table 5).
| Category | Gene | Locus | Promoter length (bp) | Pathology | Methylation |
|---|---|---|---|---|---|
| Phase I Drug Metabolism Genes | ALDH1A2 | 15q21.3 | 982 | prostate cancer | Hypermethylated |
| CYP1A1 | 15q24.1 | 1200 | head and neck cancer prostate cancer fetal growth restriction (toxics) smoking-related |
Hypermethylated Hypermethylated Hypomethylated Hypomethylated | |
| CYP1B1 | 2p22.2 | 1193 | colorectal cancer prostate cancer hepatoma cell lines breast cancer |
Hypermethylated Hypomethylated Hypermethylated Hypermethylated | |
| CYP24A1 | 20q13 | 945 | vitamin D deficiency tumor-derived endothelial cells |
Hypermethylated Hypermethylated | |
| CYP27B1 | 12q14.1 | 917 | breast cancer choriocarcinoma lymphoma and leukemia | Hypermethylated Hypermethylated Hypermethylated | |
| CYP2A13 | 19q13.2 | 928 | head and neck cancer | Hypermethylated | |
| CYP2C19 | 10q24 | 1048 | Drug resistance | Hypermethylated | |
| CYP2E1 | 10q26.3 | 918 | Parkinson's disease toluene exposure |
Hypomethylated Hypomethylated | |
| CYP2R1 | 11p15.2 | 1026 | vitamin D deficiency | Hypermethylated | |
| CYP2W1 | 7p22.3 | 934 | colorectal cancer bladder, breast, thyroid cancer liver, stomach cancer | Hypomethylated Hypomethylated Hypomethylated | |
| CYP7B1 | 8q21.3 | 1052 | prostate cancer | Hypomethylated | |
| Phase II Drug Metabolism Genes | GSTM1 | 1p13.3 | 900 | head and neck cancer | Hypermethylated |
| GSTP1 | 11q13 | 958 | toluene exposure hepatoma cells prostate cancer breast cancer |
Hypomethylated Hypermethylated Hypermethylated Hypomethylated | |
| NAT1 | 8p22 | 2132 | breast cancer | Hypomethylated | |
| SULT1A1 | 16p12.1 | 1086 | breast cancer | Hypermethylated | |
| UGT3A2 | 5p13.2 | 1076 | hepatoma cells | Hypermethylated | |
| Phase III Transporter Genes | ABCA7 | 19p13.3 | 967 | Alzheimer's disease | Hypomethylated |
| ABCB1 | 7q21.12 | 906 | breast cancer resistance to chemotherapy | Hypermethylated Hypomethylated | |
| ABCC6 | 16p13.1 | 975 | bladder cancer | Hypermethylated | |
| ABCG2 | 4q22 | 1199 | T-cell acute lymphoblastic leukemia cell lines | Hypomethylated | |
| SLC19A1 | 21q22.3 | 1040 | CNS lymphomas | Hypomethylated | |
| SLC22A3 | 6q25.3 | 1034 | prostate cancer | Hypermethylated | |
| SLC24A4 | 14q32.12 | 1029 | Alzheimer's disease | Hypomethylated |
Phase I:ALDH1A2: aldehyde dehydrogenase 1 family member A2; CYP1A1: cytochrome P450 family 1 subfamily A member 1; CYP1B1: cytochrome P450 family 1 subfamily B member 1; CYP24A1: cytochrome P450 family 24 subfamily A member 1; CYP27B1: cytochrome P450 family 27 subfamily B member 1; CYP2A13: cytochrome P450 family 2 subfamily A member 13; CYP2C19: cytochrome P450 family 2 subfamily C member 19; CYP2E1: cytochrome P450 family 2 subfamily E member 1; CYP2R1: cytochrome P450 family 2 subfamily R member 1; CYP2W1: cytochrome P450 family 2 subfamily W member 1; CYP7B1: cytochrome P450 family 7 subfamily B member 1. Phase II:GSTM1: glutathione S-transferase mu 1; GSTP1: glutathione S-transferase pi 1; NAT1: N-acetyltransferase 1 (arylamine N-acetyltransferase); SULT1A1: sulfotransferase family 1A member 1; UGT3A2: UDP glycosyltransferase 3 family, polypeptide A2. Phase III:ABCA7: ATP binding cassette subfamily A member 7; ABCB1: ATP binding cassette subfamily B member 1; ABCC6: ATP binding cassette subfamily C member 6; ABCG2: ATP binding cassette subfamily G member 2 (Junior blood group); SLC19A1: solute carrier family 19 (folate transporter), member 1; SLC22A3: solute carrier family 22 (organic cation transporter), member 3; SLC24A4: solute carrier family 24 (sodium/potassium/calcium exchanger), member 4.
Table 5: Methylation patterns in genes associated with Phase I-II drug metabolism and transporters.
Epigenetic modifications are also associated with drug resistance [116-119]. The acquisition of drug resistance is tightly regulated by post-transcriptional regulators such as RNA-binding proteins (RBPs) and miRNAs, which change the stability and translation of mRNA encoding factors involved in cell survival, proliferation, epithelialmesenchymal transition, and drug metabolism [116,117].
Conclusions
(i) Epigenetic changes (DNA methylation, histone remodeling, miRNA regulation) are common phenomena in brain disorders.
(ii) Genes associated with the pathogenesis of neurodegeneration in Alzheimer’s disease exhibit epigenetic changes suggesting that epigenetics might contribute to the pathogenesis of dementia.
(iii) DNA methylation influence phenotype differences, such as susceptibility to certain diseases and pathogens, and response to drugs and xenobiotic agents.
(iv) Epigenetic modifications are associated with drug resistance.
(v) Epigenetic modifications are reversible and can be potentially targeted by pharmacological and dietary interventions.
(vi) Epigenetic drugs can reverse epigenetic changes in gene expression and might open future avenues for the treatment of brain disorders.
(vii) A series of epigenetic drugs have been developed, including DNA methyltransferase inhibitors (nucleoside analogs, small molecules, bioproducts, antisense oligonucleotides, miRNAs), histone deacetylase inhibitors (short-chain fatty acids, hydroxamic acids, cyclic peptides, benzamides, ketones, sirtuin inhibitors, sirtuin activators), histone acetyltransferase modulators, histone methyltransferase inhibitors, histone demethylase inhibitors, and non-coding RNAs (miRNAs) with potential effects against major problems of health. Some epigenetic drugs have been approved for the treatment of different modalities of cancer.
(viii) Pharmacoepigenomics deals with the influence that epigenetic alterations may exert on genes involved in the pharmacogenomic network responsible for the pharmacokinetics and pharmacodynamics of drugs (efficacy and safety), as well as the effects that drugs may have on the epigenetic machinery.
(ix) Genes involved in the pharmacogenomic process include pathogenic, mechanistic, metabolic, transporter, and pleiotropic genes which are susceptible to epigenetic modifications leading to altered expression of proteins and enzymes, with the consequent effects on the therapeutic outcome.
(x) Although the information available at present on the pharmacoepigenomics of most drugs is very limited, growing evidence indicates that epigenetic changes are determinant in the pathogenesis of many medical conditions and in drug response and drug resistance; consequently, pharmacoepigenetic studies should be incorporated in the future as routine procedures for the proper evaluation of efficacy and safety issues in drug development and clinical trials.
References
- Cacabelos R, Fernandez-Novoa L, Lombardi V, Kubota Y, Takeda M (2005) Molecular genetics of Alzheimer's disease and aging. Methods Find Exp Clin Pharmacol 27 Suppl A: 1-573.
- Cacabelos R (2008) Pharmacogenomics in Alzheimer's disease. Methods Mol Biol 448: 213-357.
- Cacabelos R, Cacabelos P, Torrellas C, Tellado I, Carril JC (2014) Pharmacogenomics of Alzheimer's disease: novel therapeutic strategies for drug development. Methods Mol Biol 1175: 323-556.
- Szulwach KE, Jin P (2014) Integrating DNA methylation dynamics into a framework for understanding epigenetic codes. Bioessays 36: 107-117.
- Dambacher S, de Almeida GP, Schotta G (2013) Dynamic changes of the epigenetic landscape during cellular differentiation. Epigenomics 5: 701-713.
- Verhoeven KJ, Preite V (2014) Epigenetic variation in asexually reproducing organisms. Evolution 68: 644-655.
- Gavery MR, Roberts SB (2013) Predominant intragenic methylation is associated with gene expression characteristics in a bivalve mollusc. PeerJ 1: e215.
- Iacobazzi V, Castegna A, Infantino V, Andria G (2013) Mitochondrial DNA methylation as a next-generation biomarker and diagnostic tool. Mol Genet Metab 110: 25-34.
- Wang J, Yu JT, Tan MS, Jiang T, Tan L (2013) Epigenetic mechanisms in Alzheimer's disease: implications for pathogenesis and therapy. Ageing Res Rev 12: 1024-1041.
- Li LC (2013) Chromatin remodeling by the small RNA machinery in mammalian cells. Epigenetics 9: 45-52.
- Mahgoub M, Monteggia LM (2013) Epigenetics and psychiatry. Neurotherapeutics 10: 734-741.
- Kubota T, Takae H, Miyake K (2012) Epigenetic mechanisms and therapeutic perspectives for neurodevelopmental disorders. Pharmaceuticals (Basel) 5: 369-383.
- Itzykson R, Fenaux P (2013) Epigenetics of myelodysplastic syndromes. Leukemia 28: 497-506.
- Kelly TK, De Carvalho DD, Jones PA (2010) Epigenetic modifications as therapeutic targets. Nat Biotechnol 28: 1069-1078.
- Cacabelos R, Torrellas C (2014) Epigenetic drug discovery for Alzheimer's disease. Expert Opin Drug Discov 9: 1059-1086.
- Cacabelos R, Torrellas C, López-Muñoz F (2014) Epigenomics of Alzheimer’s disease. J Exp Med 6:75-82.
- Cacabelos R (2014) Epigenomic networking in drug development: from pathogenic mechanisms to pharmacogenomics. Drug Dev Res 75: 348-365.
- Harony-Nicolas H, Mamrut S, Brodsky L, Shahar Gold H, Barki Harrington, et al. (2014) Brain region-specific methylation in the promoter of the murine oxytocin receptor gene is involved in its expression regulation. Psychoneuroendocrinology 39:121-131.
- Rodrigues HF, Souza TA, Ghiraldini FG, Mello ML, Moraes AS (2014) Increased age is associated with epigenetic and structural changes in chromatin from neuronal nuclei. J Cell Biochem 115: 659-665.
- Zhao YQ, Jordan IK, Lunyak VV (2013) Epigenetics components of aging in the central nervous system. Neurotherapeutics 10: 647-663.
- Alyea RA, Gollapudi BB, Rasoulpour RJ (2014) Are we ready to consider transgenerational epigenetic effects in human health risk assessment? Environ Mol Mutagen 55: 292-298.
- Dauncey MJ (2013) Genomic and epigenomic insights into nutrition and brain disorders. Nutrients 5: 887-914.
- Rangasamy S, D'Mello SR, Narayanan V (2013) Epigenetics, autism spectrum, and neurodevelopmental disorders. Neurotherapeutics 10: 742-756.
- Tabolacci E, Chiurazzi P (2013) Epigenetics, fragile X syndrome and transcriptional therapy. Am J Med Genet A 161A: 2797-2808.
- Kubota T, Miyake K, Hirasawa T (2013) Role of epigenetics in Rett syndrome. Epigenomics 5: 583-592.
- Girardot M, Feil R, Llères D (2013) Epigenetic deregulation of genomic imprinting in humans: causal mechanisms and clinical implications. Epigenomics 5: 715-728.
- Hannula-Jouppi K, Muurinen M, Lipsanen-Nyman M, Reinius LE, Ezer S, et al. (2014) Differentially methylated regions in maternal and paternal uniparental disomy for chromosome 7. Epigenetics 9: 351-365.
- Mbadiwe T, Millis RM (2013) Epigenetics and autism. Autism Res Treat 2013: 826156.
- Blaze J, Roth TL (2013) Exposure to caregiver maltreatment alters expression levels of epigenetic regulators in the medial prefrontal cortex. Int J Dev Neurosci 31:804-810.
- Philibert RA, Beach SR, Lei MK, Brody GH (2013) Changes in DNA methylation at the aryl hydrocarbon receptor repressor may be a new biomarker for smoking. Clin Epigenetics 5: 19.
- Lee J, Hwang YJ, Kim KY, Kowall NW, Ryu H (2013) Epigenetic mechanisms of neurodegeneration in Huntington's disease. Neurotherapeutics 10: 664-676.
- Moumné L, Betuing S, Caboche J (2013) Multiple Aspects of Gene Dysregulation in Huntington's Disease. Front Neurol 4: 127.
- Suehs BT, Davis CD, Alvir J, van Amerongen D, Pharmd NC, et al. (2013) The clinical and economic burden of newly diagnosed Alzheimer's disease in a medicare advantage population. Am J Alzheimers Dis Other Demen 28: 384-392.
- Cacabelos R (2007) Pharmacogenetic basis for therapeutic optimization in Alzheimer's disease. Mol Diagn Ther 11: 385-405.
- Cacabelos R (2008) Pharmacogenomics and therapeutic prospects in dementia. Eur Arch Psychiatry Clin Neurosci 258 Suppl 1: 28-47.
- Cacabelos R (2011) The path to personalized medicine in mental disorders. In: Ritsner MS, editor. The handbook of neuropsychiatric biomarkers, endophenotypes and genes, Netherlands, Springer, pp. 3-63.
- Qureshi IA, Mehler MF (2011) Advances in epigenetics and epigenomics for neurodegenerative diseases. Curr Neurol Neurosci Rep 11: 464-473.
- Enciu AM, Popescu BO, Gheorghisan-Galateanu A (2012) MicroRNAs in brain development and degeneration. Mol Biol Rep 39: 2243-2252.
- Bertram L, McQueen MB, Mullin K, Blacker D, Tanzi RE (2007) Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat Genet 39: 17-23.
- Cacabelos R, Martínez-Bouza R (2011) Genomics and pharmacogenomics of dementia. CNS Neurosci Ther 17: 566-576.
- Cacabelos R (2005) Pharmacogenomics and therapeutic prospects in Alzheimer's disease. Expert Opin Pharmacother 6: 1967-1987.
- Cacabelos R (2009) Pharmacogenomics and therapeutic strategies for dementia. Expert Rev Mol Diagn 9: 567-611.
- Larner AJ (2013) Presenilin-1 mutations in Alzheimer's disease: an update on genotype-phenotype relationships. J Alzheimers Dis 37: 653-659.
- Hooli BV, Kovacs-Vajna ZM, Mullin K, Blumenthal MA, Mattheisen M, et al. (2014) Rare autosomal copy number variations in early-onset familial Alzheimer's disease. Mol Psychiatry 19: 676-681.
- Zou F, Belbin O, Carrasquillo MM, Culley OJ, Hunter TA, et al. (2013) Linking protective GAB2 variants, increased cortical GAB2 expression and decreased Alzheimer's disease pathology. PLoS One 8: e64802.
- Wang YL, Tan MS, Yu JT, Zhang W, Hu N, et al. (2013) Toll-like receptor 9 promoter polymorphism is associated with decreased risk of Alzheimer's disease in Han Chinese. J Neuroinflammation 10: 101.
- Li HL, Lu SJ, Sun YM, Guo QH, Sadovnick AD, et al. (2013) The LRRK2 R1628P variant plays a protective role in Han Chinese population with Alzheimer's disease. CNS Neurosci Ther 19: 207-215.
- Takeda M, Martínez R, Kudo T, Tanaka T, Okochi M, et al. (2010) Apolipoprotein E and central nervous system disorders: reviews of clinical findings. Psychiatry Clin Neurosci 64: 592-607.
- Cacabelos R (2012) Pharmacogenomics of central nervous system (CNS) drugs. Drug Dev Res 73:461-476.
- Cacabelos R, Fernández-Novoa L, Martínez-Bouza R, Mc Kay A, Carril J, et al. (2010) Future trends in the pharmacogenomics of brain disorders and dementia: Influence of APOE and CYP2D6 variants. Pharmaceuticals 3:3040-3100.
- Phillips NR, Simpkins JW, Roby RK (2014) Mitochondrial DNA deletions in Alzheimer's brains: A review. Alzheimers Dement 10: 393-400.
- Lagouge M, Larsson NG (2013) The role of mitochondrial DNA mutations and free radicals in disease and ageing. J Intern Med 273: 529-543.
- Cacabelos R, Takeda M (2006) Pharmacogenomics, nutrigenomics and future therapeutics in Alzheimer’s disease. Drugs Fut 31: 5-146.
- Veerappan CS, Sleiman S, Coppola G (2013) Epigenetics of Alzheimer's disease and frontotemporal dementia. Neurotherapeutics 10: 709-721.
- Mastroeni D, Grover A, Delvaux E, Whiteside C, Coleman PD, et al. (2011) Epigenetic mechanisms in Alzheimer's disease. Neurobiol Aging 32: 1161-1180.
- Mill J (2011) Toward an integrated genetic and epigenetic approach to Alzheimer's disease. Neurobiol Aging 32: 1188-1191.
- Edwards TM, Myers JP (2007) Environmental exposures and gene regulation in disease etiology. Environ Health Perspect 115: 1264-1270.
- Nakagawa S, Kageyama Y (2014) Nuclear lncRNAs as epigenetic regulators-beyond skepticism. Biochim Biophys Acta 1839: 215-222.
- Van den Hove DL, Kompotis K, Lardenoije R, Kenis G, Mill J, et al. (2014) Epigenetically regulated microRNAs in Alzheimer's disease. Neurobiol Aging 35: 731-745.
- Hartley I, Elkhoury FF, Heon Shin J, Xie B, Gu X, et al. (2013) Long-lasting changes in DNA methylation following short-term hypoxic exposure in primary hippocampal neuronal cultures. PLoS One 8: e77859.
- Liesz A, Zhou W, Na SY, Hämmerling GJ, Garbi N, et al. (2013) Boosting regulatory T cells limits neuroinflammation in permanent cortical stroke. J Neurosci 33: 17350-17362.
- White AO, Wood MA (2014) Does stress remove the HDAC brakes for the formation and persistence of long-term memory? Neurobiol Learn Mem 112: 61-67.
- Gräff J, Tsai LH (2013) The potential of HDAC inhibitors as cognitive enhancers. Annu Rev Pharmacol Toxicol 53: 311-330.
- Mikaelsson MA, Miller CA (2011) The path to epigenetic treatment of memory disorders. Neurobiol Learn Mem 96: 13-18.
- Wang SC, Oelze B, Schumacher A (2008) Age-specific epigenetic drift in late-onset Alzheimer's disease. PLoS One 3: e2698.
- Hernández HG, Mahecha MF, Mejía A, Arboleda H, Forero DA (2014) Global long interspersed nuclear element 1 DNA methylation in a Colombian sample of patients with late-onset Alzheimer's disease. Am J Alzheimers Dis Other Demen 29: 50-53.
- Bollati V, Galimberti D, Pergoli L, Dalla Valle E, Barretta F, et al. (2011) DNA methylation in repetitive elements and Alzheimer disease. Brain Behav Immun 25: 1078-1083.
- Tohgi H, Utsugisawa K, Nagane Y (1999) Reduction with age in methylcytosine in the promoter region -224 approximately-101 of the amyloid precursorprotein gene in autopsy human cortex. Brain Res Mol Brain Res 70:288-292.
- Octave JN, Pierrot N, Ferao Santos S, Nalivaeva NN, Turner AJ (2013) From synaptic spines to nuclear signaling: nuclear and synaptic actions of the amyloid precursor protein. J Neurochem 126: 183-190.
- Raina A, Kaul D (2010) LXR-α genomics programmes neuronal death observed in Alzheimer's disease. Apoptosis 15: 1461-1469.
- Tohgi H, Utsugisawa K, Nagane Y (1999) The methylation status of cytosines in a gene promoter region alters with age to downregulate transcriptional activity in human cerebral cortex. Neurosci Lett 275:89-92.
- Sontag E, Nunbhakdi-Craig V, Sontag JM, Diaz-Arrastia R, Ogris E, et al. (2007) Protein phosphatase 2A methyltransferase links homocysteine metabolism with tau and amyloid precursor protein regulation. J Neurosci 27:2751-2759.
- Adwan L, Zawia NH (2013) Epigenetics: a novel therapeutic approach for the treatment of Alzheimer's disease. Pharmacol Ther 139: 41-50.
- Zhang K, Schrag M, Crofton A, Trivedi R, Vinters H, et al. (2012) Targeted proteomics for quantification of histone acetylation in Alzheimer's disease. Proteomics 12: 1261-1268.
- Ding H, Dolan PJ, Johnson GV (2008) Histone deacetylase 6 interacts with the microtubule-associated protein tau. J Neurochem 106: 2119-2130.
- Julien C, Tremblay C, Emond V, Lebbadi M, Salem N Jr, et al. (2009) Sirtuin 1 reduction parallels the accumulation of tau in Alzheimer disease. J Neuropathol Exp Neurol 68: 48-58.
- Donmez G, Wang D, Cohen DE, Guarente L (2010) SIRT1 suppresses beta-amyloid production by activating the alpha-secretase gene ADAM10. Cell 142: 320-332.
- Ryu H, Barrup M, Kowall NW, McKee AC (2008) P3-260: Epigenetic modification in a monozygotic twin with Alzheimer’s disease. Alzheimers Dement 4:T598.
- Ogawa O, Zhu X, Lee HG, Raina A, Obrenovich ME, et al. (2003) Ectopic localization of phosphorylated histone H3 in Alzheimer's disease: a mitotic catastrophe? Acta Neuropathol 105: 524-528.
- Myung NH, Zhu X, Kruman II, Castellani RJ, Petersen RB, et al. (2008) Evidence of DNA damage in Alzheimer disease: phosphorylation of histone H2AX in astrocytes. Age (Dordr) 30: 209-215.
- Francis YI, Fà M, Ashraf H, Zhang H, Staniszewski A, et al. (2009) Dysregulation of histone acetylation in the APP/PS1 mouse model of Alzheimer's disease. J Alzheimers Dis 18: 131-139.
- Walker MP, LaFerla FM, Oddo SS, Brewer GJ (2013) Reversible epigenetic histone modifications and Bdnf expression in neurons with aging and from a mouse model of Alzheimer's disease. Age (Dordr) 35: 519-531.
- Liu R, Lei JX, Luo C, Lan X, Chi L, et al. (2012) Increased EID1 nuclear translocation impairs synaptic plasticity and memory function associated with pathogenesis of Alzheimer's disease. Neurobiol Dis 45:902-912.
- Koshibu K, Gräff J, Beullens M, Heitz FD, Berchtold D, et al. (2009) Protein phosphatase 1 regulates the histone code for long-term memory. J Neurosci 29: 13079-13089.
- Peleg S, Sananbenesi F, Zovoilis A, Burkhardt S, Bahari-Javan S, et al. (2010) Altered histone acetylation is associated with age-dependent memory impairment in mice. Science 328: 753-756.
- Fischer A, Sananbenesi F, Wang X, Dobbin M, Tsai LH (2007) Recovery of learning and memory is associated with chromatin remodelling. Nature 447: 178-182.
- Guan JS, Haggarty SJ, Giacometti E, Dannenberg JH, Joseph N, et al. (2009) HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 459: 55-60.
- Gräff J, Rei D, Guan JS, Wang WY, Seo J, et al. (2012) An epigenetic blockade of cognitive functions in the neurodegenerating brain. Nature 483: 222-226.
- Kim MS, Akhtar MW, Adachi M, Mahgoub M, Bassel-Duby R, et al. (2012) An essential role for histone deacetylase 4 in synaptic plasticity and memory formation. J Neurosci 32: 10879-10886.
- Ishimaru N, Fukuchi M, Hirai A, Chiba Y, Tamura T, et al. (2010) Differential epigenetic regulation of BDNF and NT-3 genes by trichostatin A and 5-aza-2'-deoxycytidine in Neuro-2a cells. Biochem Biophys Res Commun 394: 173-177.
- Tian F, Marini AM, Lipsky RH (2010) Effects of histone deacetylase inhibitor Trichostatin A on epigenetic changes and transcriptional activation of Bdnf promoter 1 by rat hippocampal neurons. Ann N Y Acad Sci 1199:186-193.
- Gupta S, Kim SY, Artis S, Molfese DL, Schumacher A, et al. (2010) Histone methylation regulates memory formation. J Neurosci 30: 3589-3599.
- Lithner CU, Hernandez CM, Nordberg A, Sweatt JD (2009)Epigenetic changesrelated to beta-amyloid-implications for Alzheimer’s disease. Alzheimers Dement 5: P304.
- Wu P, Zuo X, Deng H, Liu X, Liu L, et al. (2013) Roles of long noncoding RNAs in brain development, functional diversification and neurodegenerative diseases. Brain Res Bull 97: 69-80.
- Kumar P, Dezso Z, MacKenzie C, Oestreicher J, Agoulnik S, et al. (2013) Circulating miRNA biomarkers for Alzheimer's disease. PLoS One 8: e69807.
- Leidinger P, Backes C, Deutscher S, Schmitt K, Mueller SC, et al. (2013) A blood based 12-miRNA signature of Alzheimer disease patients. Genome Biol 14: R78.
- Alexandrov PN, Dua P, Hill JM, Bhattacharjee S, Zhao Y, et al. (2012) microRNA (miRNA) speciation in Alzheimer's disease (AD) cerebrospinal fluid (CSF) and extracellular fluid (ECF). Int J Biochem Mol Biol 3: 365-373.
- Mallick B, Ghosh Z (2011) A complex crosstalk between polymorphic microRNA target sites and AD prognosis. RNA Biol 8: 665-673.
- Nebbioso A, Carafa V, Benedetti R, Altucci L (2012) Trials with 'epigenetic' drugs: an update. Mol Oncol 6: 657-682.
- Cuadrado-Tejedor M, Oyarzabal J, Lucas MP, Franco R, García-Osta A (2013) Epigenetic drugs in Alzheimer's disease. Biomol Concepts 4: 433-445.
- Konsoula Z, Barile FA (2012) Epigenetic histone acetylation and deacetylation mechanisms in experimental models of neurodegenerative disorders. J Pharmacol Toxicol Methods 66: 215-220.
- Helin K, Dhanak D (2013) Chromatin proteins and modifications as drug targets. Nature 502: 480-488.
- Peedicayil J (2013) Epigenetic Drugs in Cognitive Disorders. Curr Pharm Des.
- Chuang DM, Leng Y, Marinova Z, Kim HJ, Chiu CT (2009) Multiple roles of HDAC inhibition in neurodegenerative conditions. Trends Neurosci 32: 591-601.
- Gapp K, Woldemichael BT1, Bohacek J1, Mansuy IM2 (2014) Epigenetic regulation in neurodevelopment and neurodegenerative diseases. Neuroscience 264: 99-111.
- Lötsch J, Schneider G, Reker D, Parnham MJ, Schneider P, et al. (2013) Common non-epigenetic drugs as epigenetic modulators. Trends Mol Med 19: 742-753.
- Cohen JE, Lee PR, Chen S, Li W, Fields RD (2011) MicroRNA regulation of homeostatic synaptic plasticity. Proc Natl Acad Sci U S A 108: 11650-11655.
- Chen S, Ge X, Chen Y, Lv N, Liu Z, et al. (2013) Advances with RNA interference in Alzheimer's disease research. Drug Des Devel Ther 7: 117-125.
- Fang M, Wang J, Zhang X, Geng Y, Hu Z, et al. (2012) The miR-124 regulates the expression of BACE1/β-secretase correlated with cell death in Alzheimer's disease. Toxicol Lett 209: 94-105.
- Zhu HC, Wang LM, Wang M, Song B, Tan S, et al. (2012) MicroRNA-195 downregulates Alzheimer's disease amyloid-β production by targeting BACE1. Brain Res Bull 88: 596-601.
- Kim IW, Han N, Burckart GJ, Oh JM (2014) Epigenetic changes in gene expression for drug-metabolizing enzymes and transporters. Pharmacotherapy 34: 140-150.
- Cacabelos R (2012) The metabolomics paradigm of pharmacogenomics in complex disorders. Metabolomics 2:1000e119.
- Cacabelos R (2012) World Guide for Drug Use and Pharmacogenomics. Corunna, EuroEspes Publishing Co.
- Cacabelos R (2015) Pharmacoepigenomics and the metabolomics of drug efficacy and safety. Metabolomics 5:e140.
- Tang X, Chen S (2015) Epigenetic Regulation of Cytochrome P450 Enzymes and Clinical Implication. Curr Drug Metab 16: 86-96.
- Kang H, Kim C, Lee H, Kim W, Lee EK (2013) Post-transcriptional controls by ribonucleoprotein complexes in the acquisition of drug resistance. Int J Mol Sci 14: 17204-17220.
- Cacabelos R, Torrellas C (2015) Epigenetics of Aging and Alzheimer's Disease: Implications for Pharmacogenomics and Drug Response. Int J Mol Sci 16: 30483-30543.
- Cacabelos R (2015) Metabolomics of drug resistance in cancer: The Epigenetic Component. Metabolomics 5:1000e141.
- Cacabelos R, Torrellas C, Carrera I (2015) Opportunities in Pharmacogenomics for the treatment of Alzheimer’s Disease. Future Neurology 10:229-252.
Relevant Topics
- Advanced Parkinson Treatment
- Advances in Alzheimers Therapy
- Alzheimers Medicine
- Alzheimers Products & Market Analysis
- Alzheimers Symptoms
- Degenerative Disorders
- Diagnostic Alzheimer
- Parkinson
- Parkinsonism Diagnosis
- Parkinsonism Gene Therapy
- Parkinsonism Stages and Treatment
- Stem cell Treatment Parkinson
Recommended Journals
Article Tools
Article Usage
- Total views: 15228
- [From(publication date):
April-2016 - Apr 02, 2025] - Breakdown by view type
- HTML page views : 14087
- PDF downloads : 1141