Effects of Immunomodulatory Peptides Derived from a Soil Bacterium on Cecal Microbiota of Broilers Challenged with Clostridium perfringens
Received: 08-Mar-2021 / Manuscript No. JIDT-22-56591 / Editor assigned: 11-Mar-2022 / PreQC No. JIDT-22-56591(PQ) / Reviewed: 22-Mar-2022 / QC No. JIDT-22-56591 / Revised: 24-Mar-2022 / Manuscript No. JIDT-22-56591(R) / Published Date: 31-Mar-2022
Abstract
Brevibacillus texasporus peptide (BT peptide) is immunomodulatory in poultry as a feed additive to substitute antibiotics. In the present study we performed 16S rRNA gene sequencing to compare development of the cecal microbial communities in Clostridium Perfringens-challenged broilers fed basal diet only or with either BT peptide (48 ppm) or antibiotics mixture (20 ppm bacitracin zinc and 40 ppm colistin sulfate). 240 chicks were randomly assigned into each treatment and a total of 65 cecal samples were collected at the beginning, during seven challenge days and one week post challenge. The composition of microbial communities was clearly distinguishable over time. Treatments with challenge and the antibiotics mixture were associated with increased diversity and with higher relative abundances of Alistipes sp. CHKCI003 and Faecalibacterium and lower abundance of Escherichia coli (all p<0.05). At the termination of the trial, the cecal microbiota in broilers supplemented with BT peptide was dominated by members of Bacteroidaceae. Predicted function analysis reveals significant enrichment of genes involved in ion-coupled transporters and sugar and biotin metabolism in the BT peptide treatment. Taken together, our results suggest that BT peptide and commonly used antibiotics have different influences on modulating the composition of cecal microbiota in broilers.
Keywords: BT peptide; Antibiotics; Broiler chicken; Bacteria; Clostridium perfringens challenge; Cecal microbiota; 16S rRNA sequencing
Introduction
Probiotics and its functionally valuable products have been shown to be a promising option to modulate the host’s immune system and suggested as an alternative to antibiotics for maintaining animal health [1]. Gram-positive soil bacteria, such as Streptomyces and Amycolatopsis, could produce Antimicrobial Peptides (AMPs), and some peptides encrypted in the microbial metaproteome act as natural effectors of the innate immune response [2,3]. Brevibacillus spp., established in 1996 and formerly known as Bacillus brevis cluster [4], are rod-shaped Gram-positive bacteria and most of them are strict aerobes. Brevibacillus spp. are widely spread in nature and some strains have been applied as probiotics for a long time [5]. It is well documented that members of the genus Brevibacillus produce antibacterial, antifungal and anti-invertebrate agents and could be a source of diverse enzymes of great biotechnological interests [6]. One group of these metabolites, Brevibacillus AMPs are synthesized through ribosomal or nonribosomal pathway, and the latter involves Nonribosomal Peptide Synthetases (NRPSs). It is interesting to note that the majority of currently-identified Brevibacillus AMPs are synthesized by NRPS machinery [7].
A group of small cationic AMPs, named Brevibacillus texasporus peptide (BT peptide), was isolated in 2005 from a soil bacterium B. laterosporus in the initial purpose of seeking novel antibiotics [8]. BT peptide has been found out to be biosynthesized through NRPS and purified to further determine its sequence. Subsequent studies have revealed that the 13-residue BT peptide shares high similarity of amino acid composition with the so-called nonribosomal linear lipopeptides secreted from different B. laterosporus strains, including Bogorols A-E and Brevibacillin [6,7,9]. Looking into the amino acid sequence alignment, the conserved ornithine in position 3 and lysines in positions 7 and 10 make this family of peptides cationic at physiological pH. Several evidences suggest that BT peptide has immunomodulatory properties that prime innate immunity and enhance leukocyte bactericidal activity to combat pathogenic infection when provided as a feed additive for broilers [10-12]. However, there are no studies, to our knowledge, examining the effects of BT peptide on gut microbiota composition of the host so far.
Necrotic Enteritis (NE) is a gastrointestinal disease of broiler chickens caused by Clostridium Perfringens strains [13]. Subclinical NE is characterized by poor performance with no clinical sign of the disease [14]. In this study, we applied a NE challenge model with broiler chickens to investigate the role of BT peptide in gut bacterial community structure shifts and compare it with the antibiotics commonly used for eradication of the pathogenic bacteria. High-throughput sequencing of 16S rRNA genes was performed to follow the response of the cecal microbial communities to C. perfringens challenge
Materials and Methods
Animals
All procedures involving animals were carried out according to experimental protocols approved by the Animal Ethics Committee of COFCO Nutrition and Health Research Institute (permit number: AR-15-0701). A total of 240 one-day-old male Arbor Acres broiler chickens were obtained and transferred from a commercial hatchery (Huadu Broiler Breeding, Beijing, China) and randomly distributed into three treatments, each group with 80 birds housed in individual cage compartments within a climate-controlled room. Each compartment had its own feeder, drinker, and brooding lamp for warmth, and plastic mesh was used for the cage floor. The ambient relative humidity inside the room was around 50%, and the environmental temperature was 32°C at placement being reduced with age to provide comfort throughout the study. Treatments were as follows: basal starter diet (coccidiostat and antibiotic-free, referred to as CP hereafter), 20 ppm bacitracin zinc+40 ppm colistin sulfate (AB), and 48 ppm BT peptide (BT), and the feeding period lasted 18 days. Feed and water were supplied under ad libitum strategy and the chickens were maintained on a 23-hour lighting program. The corn and soybean meal-based diets were formulated to meet NRC (1994) requirements. Composition of the diet and nutrient levels are presented in Table 1.
| Value | |
|---|---|
| Ingredient% | |
| Corn | 57.2 |
| Soybean meal, 46% CP | 34.6 |
| Soybean oil | 3.0 |
| Dicalcium phosphate | 1.7 |
| Limestone | 1.4 |
| 2% Premixa | 2.0 |
| Antioxidant | 0.1 |
| Nutrient composition | |
| Metabolizable energy, MJ/kg | 12.33 |
| Crude protein, % | 21.00 |
| Lys, % | 1.20 |
| Met, % | 0.50 |
| Ca, % | 1.05 |
| Available P, % | 0.45 |
aThe premix supplies the following per kg of complete feed: vitamin A, 12,500 IU; vitamin D3, 2,500 IU; vitamin K3, 2.65 mg; vitamin B1, 2 mg; vitamin B2, 6 mg; vitamin B12, 0.025 mg; vitamin E, 30 IU; biotin, 0.0325 mg; folic acid, 1.25 mg; pantothenic acid, 12 mg; niacin, 50 mg; copper, 8 mg; zinc, 75 mg; iron, 80 mg; manganese, 100 mg; selenium, 0.15 mg; iodine, 0.35 mg.
Table 1: Ingredients and nutrient composition of the diet (as-fed basis).
C. perfringens challenge was adapted from published methods [15] with some modifications. Briefly, a chicken C. perfringens type A field strain was obtained from the China Veterinary Culture Collection Center (Beijing, China) and cultured anaerobically in cooked meat medium (Aobox, Beijing, China) overnight at 37°C. At 4 to 10 days posthatch, all the birds were orally gavaged once per day at approximately the same time of morning with the actively growing cultures of this pathogenic C. perfringens (2.0 × 108 cfu/ml, 1.0 ml/bird). Plate count of viable C. perfringens was done by performing serial dilution of the culture in sterile saline and plated on sulfite-polymyxin-sulfadiazine agar (Aobox). The plates were incubated anaerobically at 37°C for 18-24 hours and colonies typical for C. perfringens were counted.
Sample collection
Samples were collected at afternoon for each sampling time. At 4 (start of challenge, referred to as T1 hereafter), 11 (1 d post-challenge, T2), and 17 (7 d post-challenge, T3) days posthatch, 6-8 birds from each treatment were randomly selected and humanely euthanized by cervical dislocation. Thus, 9 sampling groups were generated, namely CP1, CP2, CP3, AB1, AB2, AB3 and BT1, BT2, BT3 according to treatment and sampling sequence. Mucoid gut contents and sparse occurrence of focal necrosis in the small intestine with no obvious mortality implied a successful challenge. From each chicken, the cecum was removed and the cecal luminal contents were collected in a sterilized tube. The samples were flash frozen and stored at −80°C until further analysis.
DNA isolation and sequencing
Bacterial genomic DNA of each sample was extracted using the QIAamp Fast DNA Stool Mini Kit (Qiagen, Hilden, Germany) following the manufacturer’s instructions. DNA concentration and purity were monitored by agarose electrophoresis and determined by UV absorption analysis using NanoDrop spectrophotometer (Thermo Fisher Scientific, Waltham, USA). The V4 hypervariable region of the 16S rRNA bacterial gene (515-806) was amplified using specific primers with the barcodes with Phusion High-Fidelity PCR Master Mix (New England Biolabs, Ipswich, USA). PCR amplicons from each sample were pooled in equimolar amounts and purified using QIAquick Gel Extraction Kit (Qiagen). Sequencing libraries were generated using TruSeq DNA PCR-Free Sample Prep Kit (Illumina, San Diego, USA) and assessed by Qubit 2.0 Fluorometer (Thermo Fisher Scientific) and Agilent 2100 Bioanalyzer system according to the standard protocols. The final library was paired-end sequenced at 2 × 250 bp on the Illumina HiSeq 2500 platform. The raw sequence data are available at the NCBI Sequence Read Archive (SRA) database (Accession Number: SRP144400).
Sequence analyses
Raw sequences were demultiplexed and quality-filtered using QIIME (version 1.7.0) [16] to eliminate all low quality sequence reads under specific filtering conditions [17]. The resulting trimmed sequences were filtered to remove singleton reads and then grouped into Operational Taxonomic Units (OTUs) with 97% identity threshold using the USEARCH software based on the UPARSE algorithm (version 7.1) [18] while chimeric sequences were identified and removed using UCHIME [19]. The taxonomy of OTU-representative sequences was analyzed by the RDP Classifier (version 2.2) [20] against the Silva (SSU128) 16S rRNA database using confidence threshold of 70%. The resulting OTU table was used to determine taxonomic relative abundances of each sample.
Statistical analyses
Data analyses were performed with SPSS Statistics software (version 24.0, IBM Corporation, Armonk, USA) and R (version 3.3.1). Non-parametric statistical methods were used to analyze the data sets when they don’t follow the normal distribution. Differences in α-diversity for the cecal microbiota of broilers were tested using Kruskal-Wallis H test with Dunn's post hoc test and Bonferroni correction for multiple comparisons. Differences in microbiota composition as assessed by β-diversity metrics were tested using analysis of similarities (ANOSIM) or permutational multivariate analysis of variance (PERMANOVA, 999 permutations) at OTU level in the R vegan package (version 2.4-4). Differences in abundance of taxa were tested using Kruskal-Wallis H test with p-values adjusted (padj) for multiple testing by the FDR procedure. Linear Discriminant Analysis Effect Size (LEfSe) [21] was performed to identify the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways differentially represented among groups.
Results
We reproduced the subclinical necrotic enteritis disease in newborn broiler chickens by treatment of 7-consecutive-day C. perfringens challenge and followed longitudinally the development in the composition of cecal microbiota (Figure 1A). Cecal digesta samples were processed and sequenced to generate 3,535,329 clean reads using the 16S rRNA gene’s V4 hypervariable region (Table S1). After OTU clustering and rarefying to 37,970 reads per sample, 1,635 OTUs were retained for downstream analyses.

Figure 1: Microbial community composition of the cecum of broiler chickens challenged with C. perfringens. (a) Scheme of sampling strategy. T1-T3 represents the three sampling
time points in this study, generating a total of nine groups for the following analyses. (b) Bar plots show the mean values of relative abundance for the most abundant phyla in the nine
sampling groups. Only phyla present in at least 1% of the samples are shown separately.
Impact of treatments and C. perfringens challenge on composition profile and alpha diversity of the bacterial community
The cecal microbiota profiles of challenged broilers at phylum level are shown in Figure 1B. It can be observed that the phylum Proteobacteria was highly abundant in the T1 samples of all the three treatments but declined to a small portion in the T2 and T3 samples, while replaced by another phylum Bacteroidetes in these samples.
Next, alpha diversity was calculated to describe the within-sample bacterial richness and diversity using observed OTUs and Shannon index. When examining within different time points, we found that AB1 group had a significantly higher number of observed OTUs than CP1 in T1 sampling time (p=0.037, Figure 2A). We also compared alpha diversity indices in different treatments to evaluate the impact of challenge. It is shown that both bacterial community richness (p=0.018, Figure 2B) and diversity (p=0.002, Figure 2C) were notably increased over time in CP treatment. Moreover, Shannon index in AB treatment also exhibited a gradual increase during the entire course of treatment (p=0.029, Figure 2D).

Figure 2: Differences in bacterial community richness and diversity between treatments and sampling times. The bacterial abundance was represented by the numbers of OTUs and compared showing impact of treatment in T1 sampling time (a) or impact of challenge in CP treatment (b). Shannon index was used to estimate the diversity of the cecal microbiota of the challenged birds in CP (c) or AB (d) treatment [*p<0.05, **p<0.01].
Cecal bacterial community composition changed over time in challenged broilers
Bacterial community composition was significantly different between sampling groups formed by treatments and time when measured by ANOSIM distances (weighted UniFrac, r=0.514, p=0.001). These differences were recapitulated in two dimensional Non-Metric Multidimensional Scaling Analysis (NMDS) of the data based on weighted UniFrac similarity distance, which reveals a clear cut grouping structure of taxonomic composition over sampling time (Figure 3A). We then investigated the cecal microbiota at the genus level to identify the 30 most abundant taxa (Figure 3B). The heat map graphically shows that the cecal bacterial community in the nine sampling groups varied, especially observed in the course of time, which supports the ANOSIM and NDMS analyses.

Figure 3: Changes in cecal bacterial community composition over time in the broilers challenged with C. perfringens. (a) NMDS ordination based on weighted UniFrac similarity at T1 (red), T2 (blue) and T3 (green) sampling times. (b) Heatmap based on the relative abundance of thirty most abundant genera in the nine sampling groups. Clustering of groups (top) and taxa (left) is defined by the average pairwise Euclidean distance.
To single out the contribution of individual OTUs to the dissimilarity among samples, we assessed taxonomic changes that differed significantly over time in each treatment (Figure 4). Surprisingly, the abundance of OTU284 (Escherichia coli) substantially decreased above 20-fold from T1 to T2 and T3 sampling times in both CP (padj=0.036) and AB (padj=0.049) treatments. This trend coincided with the increases in the abundances of OTU1111 (Alistipes sp. CHKCI003) and OTU1175 (Faecalibacterium) over the time course of the study. In CP treatment, OTU1092 (Ruminococcaceae) and OTU1052 (Lactobacillus) were observed to increase from T1 to T2 sampling times and then decline from T2 to T3 sampling times. In contrast, the abundance of OTU1052 decreased over time in AB treatment groups (padj=0.045), and OTU304 (LachnoClostridium) showed a similar pattern of reduced abundance (padj=0.043).

Figure 4: Bar plots identifying significant differences between mean proportions of bacterial taxa in CP (a) and AB (b) treatments over time (padj <0.05). Only OTUs at least 5% abundant in at least one group are included.
Shift in bacterial communities caused by different treatments at T3 sampling time
To assess the impact of different treatments on the cecal microbial community of challenged broiler chickens, Bray-Curtis similarity-based Principal Coordinate Analysis (PCoA) was performed on sequencing data of T3 OTU level also reveal a modest global variation in bacterial composition among different treatment groups (Bray-Curtis, r2=0.222, p=0.013). We then analyzed the data using ternary plots to show the relative distribution of OTUs among T3 samples (Figure 5B). Each OTU was categorized as group representative OTUs based on whether there was a defined (10% or above) increase on its relative abundance in this treatment group compared with the other two groups. Our results suggest that CP3 and AB3 had a high relative abundance of Ruminococcaceae, whereas BT3 harbored a high relative abundance of Bacteroidaceae, which was solely contributed by OTU810 (Bacteroides dorei; see Table S2 for a complete list), and a less relative abundance of Ruminococcaceae. OTUs from the families Rikenella ceae and Porphyromonadaceae were mostly associated with the CP3 bacterial community, whereas Clostridiales_vadinBB60_group was associated with the AB3 community.

Figure 5: Taxonomic differences and predicted function of the cecal microbiota in the differently treated broilers challenged with C. perfringens at T3 sampling time. (a) PCoA plot based on Bray-Cutis distance at OTU level illustrating the distribution of microbiota by treatment group. The percentage of variation explained by the principal coordinates is
indicated on the axes. (b) Ternary plot representing the relative occurrence of individual OTUs (circles) in T3 samples. Red, blue and green circles mark OTUs at least 10% more
enriched in CP3, AB3 and BT3 than the other two groups respectively, and their relative abundance is shown as pie charts and colored by family-level taxonomic classification. Only
OTUs at least 0.5% abundant in at least one treatment are shown. (c) KEGG level 3 pathways significantly differentiated among treatments at T3 sampling time identified by LEfSe
with cutoff of linear discriminant analysis (LDA) >2. 
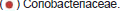
To study the functional alterations of the cecal microbiota, we used the Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) [22] to predict the functional composition profiles from the 16S rRNA sequencing data in all treatment groups at T3 sampling time. It is found that multiple KEGG level 3 categories were enriched in different treatments (Figure 5C). The pathways overrepresented in the CP3 group highlighted protein translation (ribosome, aminoacyl-tRNA biosynthesis, translation proteins, and amino acid related enzymes) and four metabolism pathways (peptidoglycan biosynthesis, energy metabolism, terpenoid backbone biosynthesis, and C5-branched dibasic acid metabolism). In contrast, the gut microbiota of the BT3 group was characterized by enrichment of other ion-coupled transporters and metabolism of sugars (galactose, fructose and mannose, and carbohydrate) as well as biotin, whereas the gut microbiota of the AB3 group was characterized by overrepresentation of benzoate degradation and isoflavonoid biosynthesis.
Discussion
The purpose of this study was to define the microbiome of broilers challenged with C. perfringens and to evaluate the impact of BT peptide on the broiler microbiome in comparison with the antibiotics mixture. Necrotic enteritis caused by C. perfringens in broilers is a contagious disease of economic importance worldwide [23]. The disease has two forms, clinical and subclinical, and most of the economic losses are related to the subclinical form and the high cost of preventing the disease with antibiotics [24]. In recent years, countries and regions where in-feed antimicrobials or antibiotic growth promoters have been banned have experienced an increase in disease outbreaks in broiler flocks [25]. Thus, it has spurred interest in investigating how it can be prevented by countermeasures other than the use of antibiotics.
BT peptide has been proposed as a feed additive for poultry to achieve effective control of food safety pathogens. It is evident that feeding the BT peptide-supplemented diet in neonatal broiler chickens induced the up-regulation of the innate immune response, reduced pathogenic bacterial colonization of the intestine and primed the cecal tissue for increased immune gene expression in response to Salmonella enterica serovar Enteritidis infection [10,11]. Researchers have realized that the large numbers of microorganisms residing in the gastrointestinal tract have a highly coevolved relationship with the host’s immune system [26], supported by the fact that disturbances in the bacterial microbiota result in dysregulation of immune cells [27]. Particularly, it is well understood that the interaction between the gut immune system and commensal microbes in chickens starts immediately after hatching [28]. Since BT peptide is not absorbed in the intestine [12], we considered the effect of its supplementation on diversity and function of the avian gut microbiota.
As described in results, the counteracting relationship of Proteobacteria and Bacteroidetes observed between T1 and T2/T3 can be partially explained by the high existence of the genus Escherichia-Shigella in the first week of age and Bacteroides thereafter, which shares strong similarity with findings of previous research [29,30]. A core bacterial microbiota of broiler cecum was proposed comprising Escherichia-Shigella at 0 and 7 days of age and Bacteroides, Erysipelotrichaceae, Faecalibacterium, Lachnospiraceae, Oscillospira, Rikenella, Ruminococcus, Streptococcus and Lactobacillus spp. after 14 days of age [29,31]. Temporal changes in the chicken cecal microbiota suggest that taxonomic richness and diversity typically increase from day of hatch to maturity [32], as revealed by our alpha diversity analysis.
The pathogenic strain that was used for challenging broiler chickens belongs to the genus Clostridium, which could be a predominant bacterium in the cecum depending on the age of the bird [33]. However, we did not detect a marked colonization of the cecum with C. perfringens throughout the sampling time. One of the possible reasons is that the gross lesions caused by C. perfringens infection usually happen in the small intestine including jejunum and ileum [24], thus in the cecal mucosa may not be the primary site for the pathogen to colonize. We also notice that a low relative abundance of Clostridium in 16S rRNA sequencing data and low cecal counts of C. perfringens were obtained in a study on broilers at 0-36 days of age [34], implying a difficult colonization of the cecal environment for C. perfringens in the early stage of broiler chickens.
Analysis of the microbiome at 7 d post-challenge shows that the most abundant organisms were discriminating among different treatments. For challenged broilers fed with BT peptides, the dominant taxa were Bacteroides dorei and Ruminococcaceae. Bacteroides colonization of the gastrointestinal tract at early stage of life is important given their role in the digestion of complex carbohydrates to fermentation products which are beneficial to hosts [35,36]. Notably, our PICRUSt results also reveal that predicted genes from the metagenome related to multiple carbohydrate metabolic pathways were significantly increased in BT treatment. Bacteroides dorei, particularly, has been found to exhibit strong association with the development of autoimmunity in type 1 diabetes in a Finnish cohort of children [35]. The structurally and functionally distinct lipopolysaccharide of Bacteroides dorei is assumed to play an important role in immune regulation in human individuals [37]. This study lacks sufficient data to define the effect of high Bacteroides abundance on broiler immune response to C. perfringens pathogen, and further studies are needed to investigate this potential link.
Various bacteria belonging to Ruminococcaceae were most abundant in both CP and AB treatments at 7 d post-challenge. Members of the family Ruminococcaceae including Faecalibacterium are known for their ability to decompose plant material and convert into butyrate and other short-chain fatty acids that can be absorbed and used for energy by the host. Significant enrichment of pathways regulating energy metabolism in CP treatment also demonstrates a heightened demand for energy by the cells of the intestine in this condition. Furthermore, we identified two Bacteroidetes OTUs, OTU1111 (Alistipes sp. CHKCI003, a species from the family of Rikenella ceae) and OTU377 (Barnesiella, a genus from the family of Porphyromonadaceae), which were more enriched in CP treatment. These two bacteria have been reported to contribute to host immune development. The appropriate proportion of Alistipes is suspected to augment intestinal immune maturation [38], and administration of the commensal Barnesiella could eradicate the infection of vancomycin-resistant Enterococcus [39], suggesting the microbiota as a required component of the effector response of the host [40]. Nevertheless, we need more research to unravel the underlying mechanism of this bacterial community development.
Conclusion
Collectively, our results show that feeding C. perfringens-challenged broilers with BT peptide or the antibiotics causes a different outcome on the composition of microbial community in cecum. Additionally, analysis of the predicted metagenomes indicates functions involved with improved sugar metabolism when supplemented with BT peptide. Future studies should include a more explicit investigation of the host-microbe interaction to depict a detailed picture of the immunomodulatory activity of BT peptide from the perspective of gut microbiota, which may benefit BT peptide as a novel antibiotics alternative for a range of livestock species.
Disclosure Statement
The authors declare that they have no competing interests.
Acknowledgements
This work was supported by the Beijing Municipal Science & Technology Program under Grant Z141100002614014; the National Key R&D Program of China under Grant 2018YFD0500600; and the Basic Research Funds of COFCO Nutrition and Health Research Institute. We appreciate Dr. Yiwei Jiang (My Galaxy LLC, Ft. Worth, USA) for providing the BT peptide material for this study. We would like to thank Dr. Yuming Guo (China Agricultural University, Beijing, China) for technical assistance with the challenge experiment and Dr. Shuangli Mi (Beijing Institute of Genomics, Chinese Academy of Sciences, Beijing, China) for assistance with sample sequencing during the study.
References
- Kanmani P (2013) Probiotics and its functionally valuable products-a review. Critical Rev Food Sci and Nut 53:641-658.
[Crossref] [Googlescholar] [PubMed]
- Lewies A, Du Plessis LH, Wentzel JF (2019) Antimicrobial peptides: The achilles‘heel of antibiotic resistance?. Probiotics Antimicrob Proteins 11:370-381.
[Crossref] [Googlescholar] [PubMed]
- Chen CH, Lu TK (2020) Development and challenges of antimicrobial peptides for therapeutic applications. Antibiotics 9.
- Panda AK (2014) Brevibacillus as a biological tool: A short review. Antonie Van Leeuwenhoek 105:623-639.
- Sanders ME, Morelli L,Tompkins TA (2003) Sporeformers as human probiotics: bacillus, sporolactobacillus, and brevibacillus. Comprehensive Rev in Food Sci and Food Safe 2: 101-110.
- Yang X (2016) Isolation and structural elucidation of brevibacillin, an antimicrobial lipopeptide from brevibacillus laterosporus that combats drug-resistant gram-positive bacteria. App Environ Microbiol 82:2763-2772.
[Crossref] [Googlescholar][PubMed]
- Yang X, Yousef AE (2018) Antimicrobial peptides produced by brevibacillus spp.: Structure, classification and bioactivity: A mini review. World J Microbiol Biotechnol 34:57.
- Wu X, Ballard J, Jiang Y (2005) Structure and biosynthesis of the bt peptide antibiotic from brevibacillus texasporus. App Environ Microbiol 71: 8519-8530.
- Barsby T (2006) The bogorol family of antibiotics: Template-based structure elucidation and a new approach to positioning enantiomeric pairs of amino acids. J Org Chem 71: 6031-6037.
- Kogut MH (2007) The effects of the BT/TAMUS 2032 cationic peptides on innate immunity and susceptibility of young chickens to extraintestinal salmonella enterica serovar enteritidis infection. Int Immunopharmacol 7: 912-919.
- Kogut MH (2010) Feeding the BT cationic peptides to chickens at hatch reduces cecal colonization by Salmonella enterica serovar Enteritidis and primes innate immune cell functional activity. Foodborne pathogens and disease 7:23-30.
[Crossref] [Googlescholar] [PubMed]
- Kogut MH (2012) BT cationic peptides: Small peptides that modulate innate immune responses of chicken heterophils and monocytes. Vet Immunol Immunopathol 145:151-158.
[Crossref] [Googlescholar] [PubMed]
- Lacey JA (2018) Clostridium perfringens-mediated necrotic enteritis is not influenced by the pre-existing microbiota but is promoted by large changes in the post-challenge microbiota. Vet Microbiol 227:119-126.
- Wang H (2017) Probiotic enhanced intestinal immunity in broilers against subclinical necrotic enteritis. Front Immunol 8:1592.
- Liu D (2010) Exogenous lysozyme influences clostridium perfringens colonization and intestinal barrier function in broiler chickens. Avian Pathol 39:17-24.
- Caporaso JG (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7: 335-336.
- Bokulich (2013) Quality-filtering vastly improves diversity estimates from illumina amplicon sequencing. Nat Methods 10:57-59.
- Edgar RC (2013) UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods 10:996-998.
- Edgar RC (2011) UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27:2194-2200.
- Wang Q (2007) Naive bayesian classifier for rapid assignment of rrna sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261-5267.
- Segata N (2011) Metagenomic biomarker discovery and explanation. Genome Biol 12: R60.
[Crossref] [Googlescholar] [PubMed]
- Langille MG (2013) Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 31:814-821.
[Crossref] [Googlescholar] [PubMed]
- Yitbarek A (2012) Innate immune response to yeast-derived carbohydrates in broiler chickens fed organic diets and challenged with Clostridium perfringens. Poultry science 91:1105-1112.
[Crossref] [Googlescholar] [PubMed]
- Shojadoost B, Vince AR, Prescott JF (2012) The successful experimental induction of necrotic enteritis in chickens by Clostridium perfringens: A critical review. Vet res 43:74.
- Van Immerseel F (2004) Clostridium perfringens in poultry: An emerging threat for animal and public health. Avian Pathol 33:537-549.
- Hooper LV, Littman DR, Macpherson AJ (2012) Interactions between the microbiota and the immune system. Science 336:1268-1273.
[Crossref]
- Round JL, Mazmanian SK (2009) The gut microbiota shapes intestinal immune responses during health and disease. Nat RevImmunol.
- Crhanova M (2011) Immune response of chicken gut to natural colonization by gut microflora and to Salmonella enterica serovar enteritidis infection. Infect Immun 79: 2755-2763.
[Crossref] [Googlescholar] [PubMed]
- Johnson TJ (2018) A consistent and predictable commercial broiler chicken bacterial microbiota in antibiotic-free production displays strong correlations with performance. App Environ Microbiol.
[Crossref] [Googlescholar] [PubMed]
- Waite DW, Taylor MW (2014) Characterizing the avian gut microbiota: membership, driving influences, and potential function. Fron Microbial 5:223.
- Stanley D, Hughes RJ, Moore RJ (2014) Microbiota of the chicken gastrointestinal tract: influence on health, productivity and disease. App Microbiol Biotechnol 98: 4301-4310.
- Oakley BB, Kogut MH (2016) Spatial and temporal changes in the broiler chicken cecal and fecal microbiomes and correlations of bacterial taxa with cytokine gene expression. Front vet Science 3:11.
- Stanley D (2012) Intestinal microbiota associated with differential feed conversion efficiency in chickens. App Microbiol Biotechnol 96:1361-1369.
- Ranjitkar S (2016) Bacterial succession in the broiler gastrointestinal tract. App Environ Microbiol 82:2399-2410.
- Davis-Richardson AG (2014) Bacteroides dorei dominates gut microbiome prior to autoimmunity in Finnish children at high risk for type 1 diabetes. Frontiers in Microbiology 5: 678.
[Crossref] [PubMed]
- Martens EC, Chiang HC, Gordon JI (2008) Mucosal glycan foraging enhances fitness and transmission of a saccharolytic human gut bacterial symbiont. Cell Host Microbe 4: 447-457.
- Vatanen (2016) Variation in microbiome lps immunogenicity contributes to autoimmunity in humans. Cell 165:842-853.
- Chung, H (2012) Gut immune maturation depends on colonization with a host-specific microbiota. Cell 149:1578-1593.
- Ubeda C (2013) Intestinal microbiota containing barnesiella species cures vancomycin-resistant enterococcus faecium colonization. Infect Immun 81: 965-973.
[Crossref] [Googlescholar] [PubMed]
- Belkaid Y, Hand TW (2014) Role of the microbiota in immunity and inflammation. Cell, 2014. 157: 121-141.
Citation: Li X, Li G, Peng Q, Sun Y, Li Y, et al. (2022) Effects of Immunomodulatory Peptides Derived from a Soil Bacterium on Cecal Microbiota of Broilers Challenged with Clostridium perfringens.J Infect Dis Ther S1:004.
Copyright: © 2022 Li X, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Share This Article
Recommended Journals
Open Access Journals
Article Usage
- Total views: 1672
- [From(publication date): 0-2022 - Feb 22, 2025]
- Breakdown by view type
- HTML page views: 1324
- PDF downloads: 348