Research Article Open Access
Development of a HPLC-UV Method for the Simultaneous Determination of Intracellular Glutathione Species in Human Cells
Lipsa D1, Cacho C2, Leva P1, Barrero-Moreno J1* and Aguar P1
1European Commission, Joint Research Centre, Institute for Health and Consumer Protection, Chemical Assessment and Testing Unit, Ispra (VA), Italy
2Analytical Chemistry Department, Faculty of Science, Palacky’s University in Olomouc, Olomouc, Czech Republic
- *Corresponding Author:
- Barrero-Moreno J
European Commission, Joint Research Centre
Institute for Health and Consumer Protection
Chemical Assessment and Testing Unit
via E. Fermi 2749, I-21027
Ispra (VA), Italy
Tel: +390332789863
E-mail: josefa.barrero-moreno@jrc.ec.europa.eu
Received date: July 02, 2015; Accepted date: July 20, 2015; Published date: July 27, 2015
Citation: Lipsa D, Cacho C, Leva P, Barrero-Moreno J, Aguar P (2015) Development of a HPLC-UV Method for the Simultaneous Determination of Intracellular Glutathione Species in Human Cells. J Anal Bioanal Tech 6:259. doi: 10.4172/2155-9872.1000259
Copyright: © 2015 Lipsa D, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Visit for more related articles at Journal of Analytical & Bioanalytical Techniques
Abstract
In the present work, an HPLC-UV method was set-up to allow the simultaneous quantification of the reduced- GSH, oxidised-GSSG and nitroso-GSNO glutathione species. Chromatographic separation was achieved on YMC ODS-A C18 column (150 × 4.6 mm, 5 μm), coupled to a Guard-c precolumn (YMC-Pack, 10 × 1-4,0 mm). The eluted compounds were detected at 215 nm by UV-detector, by keeping the column oven at room temperature while the auto-sampler temperature was maintained at 4°C. A fractional factorial design has been applied for the optimization of the mobile phase resulting in baseline separated peaks within 6 minutes. In-house validation was evaluated by linearity, limits of detection (LODs), limits of quantification (LOQs), reproducibility, repeatability and recovery. The detection and quantification limits obtained for standard solutions were below 0.2 μM and 0.6 μM, respectively (RSD values below 2%). The developed method was applied to the measurement of GSH, GSSG and GSNO in human pulmonary cells (A549) exposed to limonene, limonene oxide solubilized into the culture medium and to NO2 as gas phase. Results show an increase in GSH levels, without significant changes in GSSG, when cells were exposed to limonene oxide, while cells exposed to NO2 resulted in a significant increase of GSNO amount. Detection limits were of 1 μM for the glutathione species measured in A549 cells, with RSD values below 2.5%. In conclusion, the present HPLC-UV method can be readily used to measure in a rapid, simultaneous and accurate way the status of GSH, GSSG and GSNO in human cells, their simultaneous quantification helping to better predict the potential impact of chemicals on human health.
Keywords
HPLC-UV method; Glutathione species (GSH, GSSG, GSNO); Alveolar epithelial cell line (A549); Limonene; Limonene oxide; Nitric dioxide
Introduction
Intracellular glutathione plays a key role in cell protection since it reacts with potentially dangerous endogenous and exogenous compounds detoxifying them [1,2]. Two different mechanisms have been mentioned in the literature to describe the detoxifying properties of glutathione. On the one hand, xenobiotics can be detoxified by the formation of an intermolecular disulphide bond with gluthathione thus being reduced. On the other hand, the thiol group is able of complexing compounds having heavy metals [3,4]. Together with thioredoxin, glutathione is the most important intracellular antioxidant, reducing free radicals or peroxides and maintaining proteins, enzymes and vitamins in their reduced active state [5,6]. Additionally, glutathione traps any nitric oxide or peroxynitrite which is formed during cell metabolism to form S-nitroso-GSH [7].
In mammalian cells, the most abundant glutathione species are: 1) Reduced glutathione, GSH, which is the active species participating in cell protection; 2) Oxidized glutathione, GSSG, which is formed due to the antioxidant activity of glutathione, and therefore increases in the presence of oxidative stress caused by e.g. free radicals or peroxides; 3) S-nitroso glutathione, GSNO, which is formed by the conjugation of the thiol group in GSH with nitric oxide (NO) and peroxynitrite formed during cell metabolism [6].
In normal conditions, human hepatic cells produce the most reduced glutathione (e.g. HepG2 cells 52.9 ± 3.1 nmol mg-1 protein), while other type of cells are producing reduced glutathione to some extent (e.g. human prostate cells (DU154) 20.21 ± 2.44 nmol mg-1 protein; human differentiated macrophages cells (THP-1) 13.71 ± 3.83 nmol mg-1 protein) [8,9].
The ratio between reduced (GSH) and oxidized (GSSG) glutathione is commonly used to assess the cell toxicity of certain compounds which possess redox properties [10]. In normal cells, the optimal GSH:GSSG ratio exceeds 100, while in case of cellular oxidative damage, this ratio was reported to decrease to values between 10 and 1 [11]. Decreased levels of glutathione ratios have been linked to a number of diseases states such as cancer, HIV, diabetes, Alzheimer’s and Parkinson’s diseases etc. [12-14]. Because of its crucial role in human health status (e.g. protects the airways from chemical insult by regulating many of the bioactivities of inflammatory cells), determining the content of glutathione species in human biological samples can be of interest from the clinical point of view. In this sense, it helps understanding the mode of action of new drugs that are applied in preventing or treating cancer.
Different methods have been described in the literature to measure reduced (GSH) and oxidized glutathione (GSSG) forms in plants (e.g. cucumber leaves), in plasma/serum of animals or humans etc. [15-18]. These methods are based on ELISA tests using commercially available kits or on their separation by HPLC with fluorescence or UV detection [19-21]. ELISA kits usually require a high amount of cells (~106 cells) to do the analysis [19]. Fluorescent determination, on the other hand, generally provides a considerably higher sensitivity, but presents the drawback of requiring a derivatization step in order to form fluorescent compounds. Derivatization is normally carried out by the reaction with dansyl chloride or with o-phthalaldehyde [20-23]. This derivatization step implies longer and more complex sample treatment due to extreme reaction conditions. Additionally, the added excess reagent has to be removed after the derivatization to avoid its interference with the chromatographic analysis. Finally, the simultaneous determination of GSH and GSSG is not possible as the derivatization reaction is selective either towards thiol groups or disulphide bonds. Thus, two parallel analyses must be done in order to establish the amount of GSH and GSSG in the sample. Recently, it has been demonstrated that during derivatisation steps, in particular with o-phthalaldehyde, glutathione species are falsely detected since this compound easily reacts with glutathione or with other sulfhydryl compounds [24].
Few publications have appeared in recent years presenting an HPLC-UV method for the determination of GSH and GSSG in animal plasma samples without need of glutathione derivatization [25]. On the other hand, GSNO has been measured separately from GSH and GSSG by changing the column of the HPLC coupled with UV detection, which implies time consuming and additional costs [26].
Determination of intracellular glutathione (GSH), glutathione disulphide (GSSG) and S-nitrosoglutathione (GSNO) in human lung cells is of high relevance since the number of people affected by pulmonary disease (e.g. asthma, lung cancer) is rapidly increasing (e.g. “cancer rates could further increase by 50% to 15 million new cases in the year 2020”) [27]. Therefore, detection of changes in the antioxidant cellular defense could contribute to early identify some of the chemicals/ pollutants present in human lifestyle and which might be responsible for the illness. One of the common pollutants present in human lifestyle is R-limonene, an abundant volatile organic compound (VOC), found also in indoor environments, as it is released from various sources such as consumer products (e.g. air fresheners, cleaning products) where it is added as a fragrance [28,29]. Limonene oxide, on the other hand, can be formed during the oxidation of limonene in the presence of atmospheric ozone [30]. Nitric dioxide (NO2) is a priority (major) indoor air pollutant due to the high concentrations reached during e.g. gas heating or cooking that can seriously affect asthmatic people [31]. A concentration of 200 μg m-3 1 hour mean has been established as a safety guideline for the concentration of NO2 in indoor air in the WHO guidelines [32].
The present study describes the development and optimisation of an HPLC-UV method suitable to quantify variations in the concentration of GSH, GSSH, GSNO formed in a human carcinogenic pulmonary cell line (A549) under physiological and chemical’s exposure conditions. The proposed methodology allows cost-efficient and simultaneous quantification of the three glutathione species: GSH, GSSG and GSNO without the need of a derivatization step prior to the analysis of the selected glutathione species in the biological materials (e.g. human lung cells extracts).
Thus, following in-house validation of the HPLC-UV method, the depletion or the increase in the concentration of the three selected glutathione forms measured in A549 cells, exposed to liquid limonene, limonene oxide (solubilized into the culture media) and to the gaseous phase of NO2 (air-liquid interface), is investigated within a single chromatographic run.
Materials
Chemical reagents and materials
Trifluoroacetic acid (TFA), acetonitrile, sodium perchlorate, picric acid, metaphosphoric acid (MPA), R-limonene (liquid phase) and limonene oxide were ordered from Sigma-Aldrich (Sigma Aldrich, St. Louis MO, USA). Additionally, ammonium sulfate, glutathione (GSH), glutathione disulphide (GSSG) and S-nitrosoglutathione (GSNO) standards were purchased from Sigma-Aldrich. NO2 (gas-phase) were ordered from Air Liquide (Italy). Water deionized with a Milli-Q system (Millipore, USA) was used throughout.
The following columns were used for the HPLC-UV analysis of GSH, GSSG and GSNO: Synergy Fusion, Kinetex C18 columns bought from Phenomenex while YMC ODS-A was purchased from Agilent Technologies.
Safety considerations: Since picric acid it has been shown to be an explosive compound, safety guidelines (e.g. working under a wellventilated fume hood, wearing protective equipment etc.) were applied during all the experiments to prevent inherent dangers of picric acid use [33].
Biological reagents and materials
Roswell Park Memorial Institute medium (RPMI 1640), fetal bovine serum (FBS), penicillin-streptomycin, 0.25% trypsin/EDTA, phosphate buffered saline (PBS) and 4-2-hydroxyethyl-1-piperazinyletanesolfonic acid (HEPES) were purchased from Invitrogen (USA). Mammalian protein extraction reagent (M-PER) and bicinchoninic acid assay was purchased from Thermo Fischer (Italy).
The human cell line used is the lung epithelial carcinoma A549 which was obtained from American Type Culture Collection (ATCC #: CCL-185).
Instrumental
Exposure equipment for human lung cells: Direct exposure of the human lung cell lines (A549) at the air/liquid interface was performed by using an in vitro cell culture exposure device named CULTEX (Germany).
Chemical liquid treatment of the selected cell lines was carried out in cell culture plates under a biological fume hood (Steril-CTH, Angelantoni Life Science) and then placed in an incubator Forma Series II 3110 Water-Jacketed CO2, (Thermo Scientific, USA).
Extraction equipment: Ultrasonic water bath (Starsonic 35, 28- 34 Hz) was supplied by Liarre, Italy. A 5417R centrifuge (Eppendorf, Germany) was used to separate the supernatant from the cellular debris. Microcentrifuge tubes (Amicon filters, Millipore, USA) were used to collect, filtrate and concentrate the cellular extract. The protein quantification from the cellular debris was performed by using an EnSpire Multimode Plate Reader with integrated software (Perkin Elmer).
HPLC-UV equipment: The separation and determination of GSH, GSSG and GSNO molecules was carried out using an Agilent 1100 Series HPLC system composed of binary pump, autosampler and diode array detector (Agilent, Santa Clara, USA). Their separation was achieved on a ODS-A C18 column (YMC, Japan. YMC-Pack, 150 × 4.6 mm) with a 5 μm particle size, coupled to a Guard-c precolumn (YMCPack, 10 × 1-4,0 mm). Data were processed with ChemStation software (version A.08.03, Agilent).
Methods
Chromatographic conditions for the detection of intracellular GSH, GSSG and GSNO
The HPLC analysis was performed using isocratic elution with a mobile phase’s composition of water/acetonitrile (H2O/can: 95/5, v/v), trifluoroacetic acid (TFA: 0.1%) and sodium perchlorate (12 mg mL-1) and a flow rate that was adjusted to 1 mL min-1. The detection wavelength was set at 215 nm by UV detector. The column oven temperature was kept at room temperature while the auto-sampler temperature was maintained at 4°C. A volume of 10 μL of standards or sample solutions (e.g. the cell culture lysate) was directly injected to the HPLC equipment and further analysed. Under these conditions the three selected glutathione species were simultaneous determined in less than 6 minutes.
Different chromatographic conditions such as mobile phase ratio, injection volume, and flow rate were optimised before the validation of the method. In-house validation was evaluated by linearity, limits of detection (LODs), limits of quantification (LOQs), reproducibility, repeatability and recovery.
Preparation of the standards (GSH, GSSG, GSNO) and of the tested chemicals solutions
Stock solutions containing 20 mM GSH (MW 307.3), GSSG (MW 612.6), GSNO (MW 336.3), were prepared in the same buffer as was used for the intracellular extraction of glutathione (e.g. 0.5% picric acid+25 mM ammonium sulfate added to the mobile phase).
The tested liquid chemicals (e.g. limonene and limonene oxide) were dissolved at their maximum solubility in RPMI 1640 medium containing 1% FBS. After well mixing, the initial stock solution of R-limonene and limonene oxide (+) was further diluted in RPMI 1640 containing 1% FBS and their subsequent concentrations (e.g. R-limonene: 0; 3.45 mg L-1; 6.9 mg L-1; 13.8 mg L-1) were tested for their oxidant capacity in A549 cells.
Stability tests
Glutathione stability in standards solutions and biological samples: In order to avoid decomposition or oxidation of glutathione molecules, aliquots of the standard solutions (GSH, GSSG, GSNO) were stored at -80°C for 6 months (weakly verification tests were performed by quantifying diluted aliquots of selected glutathione species).
Previous studies indicate that glutathione can decompose due to enzymatic degradation and/or photolysis [34]. Thus, dark amber vials were used for both standards and samples storage and analysis with the scope to prevent that direct light induce the degradation of glutathione (e.g. the polar covalent bond between sulphur and nitrogen is susceptible to homolysis by direct light) [35]. Moreover, the limitation of enzymatic decomposition of glutathione content present in biological samples was achieved by keeping the samples on ice during the extraction procedure and by storing the cellular extract samples at -80°C for 2 months.
During their analysis within the same day, standard and samples solutions showed to be stable when were kept and cooled to 4°C on the auto-sampler well plate (stability tests were carried out after 8 h considering these storage conditions).
Target chemicals stability: The potential interaction of selected chemicals with the cellular medium components (e.g. protein) was investigated before running any in vitro toxicological test. The interaction of the test chemicals with the culture medium would lead to an uncontrolled environment exposure for cells, since the target chemical concentration would be continuously modified over time. Therefore, qualitative tests were prepared and performed using an Agilent GC 6890 with a liquid injection auto-sampler for the evaluation of the stability of limonene and limonene oxide solubilized into cell culture medium. Target chemicals were solubilized at a concentration of 0.6 mg mL-1 into the culture medium, which was containing two different concentrations of FBS: one with 10% and the other with 1%. Stability of limonene and limonene oxide prepared in culture medium was obtained when only 1% FBS was added to RPMI 1640 (data not shown here). Therefore, the oxidant capacity of target liquid chemicals was tested in A549 cells under the mentioned culture medium composition.
Cell culture maintenance
A549 cells were grown in T-75 cm2 culture flasks using an RPMI 1640 medium with 25 mM HEPES, supplemented with 10% fetal bovine serum, 100 μg mL-1 penicillin and streptomycin at 37°C under a humidified atmosphere containing 5% CO2. The medium was changed every 2 days. Cells were used between passages 10 and 12. Once the near-confluent state was reached, the A549 cells were washed with PBS, and then harvested by the addition of 0.25% trypsin for 5 minutes. The number of cells was determined using the automatic cell counter, Scepter 2.0 [36].
Cell exposure methods
Conventional culture conditions were applied for the exposure of A549 cells to R-limonene and its oxidized form, limonene oxide (+). Cells were seeding at a culture density of 100,000 cells/well and grown as a monolayer in Falcon 24 well-plate at 37°C and 5% CO2. After reaching 90-100% confluence, cells were incubated with varying concentrations of limonene or limonene oxide for 24 hours. Accordingly, cells were incubated with concentrations as high as 13.8 mg L-1 and 137 mg L-1 of R-limonene and limonene oxide (+) respectively, corresponding to their maximum solubility in the culture media at room temperature.
For the exposure experiments of cell culture to NO2, cells were seeded onto porous transwell membranes (A549 cells: 300,000 cells/ well) with a pore diameter of 0.4 μm (Falcon, PET membrane) and were directly exposed to a NO2 gas-phase by the use of an air-lifted interface exposure system, called CULTEX. The cells were thus fumigated for 1 and 2 hours with a flow of clean air containing 12 ppm of NO2 at a flow of 2 mL min-1 as described in detail elsewhere [37]. A comparable set of transwells cultivated under conventional conditions (e.g. 5% CO2) and another one exposed to synthetic air were used as negative controls.
Under both exposure conditions (test chemicals solubilized into the culture media and air-lifted cultures) immediately after exposure, the cells were prepared for the extraction of glutathione species. For this purpose, the cellular extracts were directly injected into HPLC-UV or stored at -80°C.
Extraction procedure of GSH, GSSG, GSNO species from A549 cells
After the different chemical exposures with the cell culture (A549), the following steps were executed with the scope to further determine the three intracellular glutathione species:
(i) culture medium was discarded from the cells exposed to the test chemicals solubilized into the culture media; (ii) Cells were washed twice with cold PBS (200 μL for 24-well plate and 300 μL for 6 well plate); (iii) 300 μL or 600 μL of ammonium sulfate (25 mM) dissolved in 0.5% picric acid - prepared in mobile phase - was added to the cells (during this step, cells were kept on ice); (iv) After incubation at -18°C for 10 minutes, the cellular homogenates were mechanically scraped for approximately 5 minute and transferred to an Amicon 3K Ultra Millipore filter (v) the cellular suspension was sonicated in icy water for 2 minutes (vi) then it was centrifuged for 15 minutes at 4°C (12000 g) in order to remove proteins and high molecular weight compounds prior to HPLC analysis (vii) finally, the supernatant was immediately transferred to a pre-chilled dark amber vials and injected into the HPLC or stored at -80°C for further analysis of GSH, GSSG and GSNO.
Determination of protein content
The pellet (containing proteins) was collected from the Amicon 3K Ultra Millipore filter and quantified by bicinchoninic acid assay (BCA assay) according to the manufacturer's recommended protocol.
Cellular viability by neutral red uptake assay
Prior to the determination of the possible oxidant activity induced by limonene, limonene oxide and nitric dioxide on A549 cells, the identification of chemical’s concentration in which no statistically significant effect on the loss of cell viability when compared to untreated cells was investigated. Therefore, the concentration and exposure time dependence of limonene, limonene oxide and nitric dioxide on cellular viability was assessed by running a neutral red uptake assay (NRU) [38]. Once the A549 cells were exposed to the tested chemicals, the neutral red dye was added to the cells, and then the cells were incubated for 3 hours at 37°C. Afterwards, the supernatant was aspirated and the plates were washed with PBS, followed by the addition of an acetic acid/ water/ethanol (1:49:50, v/v/v) solution. Plates were allowed to incubate for 30 minutes at room temperature prior to the measurement of the absorbance at 540 nm.
In-house validation of the proposed method
Linearity: Method linearity was evaluated by the construction of calibration curves using standard solutions in the ranges of 2.5 to 370 μg mL-1. Calibration curves were obtained by plotting the known concentration of GSH, GSSG or GSNO against the area units of the peaks. Each injection was done in 5 replicates. The concentrations of GSH, GSSG and GSNO in unknown cellular extracts were calculated based on the regression equations obtained.
Limits of detection and quantification: The limits of detection (LOD) were calculated as three times the signal/noise (S/N) average, while the limits of quantification were defined as the amounts giving an S/N ratio of 10. They were expressed in terms of concentration by means of the standard calibration curve for each target compound.
Accuracy and precision: The accuracy of the method was established based on the percent recovery study conducted in the supernatant of the biological samples spiked with two different concentrations of the GSH, GSSG and GSNO standard solutions.
Precision was evaluated both in terms of repeatability and reproducibility of the method. The repeatability of the method (evaluated by the RSD) was assessed from four concentrations of each of the glutathione species and its four independent replicates. Long term reproducibility of the method (2 months) was assessed by the RSD from four different concentrations of freshly prepared standard solutions.
Results and Discussion
Optimization of the HPLC mobile phase used to separate GSH, GSSG and GSNO
A fractional factorial design was considered for the optimization of the mobile phase composition used in the chromatographic separation of GSH, GSSG and GSNO. A set of sixteen combinations of the four selected mobile phase compounds (water, acetonitrile, trifluoroacetic acid and sodium perchlorate) was carefully chosen to be tested in a practical way according to the ranges specified in Table 1. The mobile phases to be tested were selected to ensure a representative model of all the possible combinations of their four main constituents: water, acetonitrile, trifluoroacetic acid and sodium perchlorate (e.g. 1st combination tested was consisting in water (80%): acetonitrile (20%): trifluoroacetic acid (0%): sodium perchlorate (0 mg mL-1) (Table 1).
| Tested mobile phase composition composed | ||||
|---|---|---|---|---|
| H2O (%) | 80 | 90 | 95 | 100 |
| Acetonitrile (%) | 20 | 10 | 5 | 0 |
| TFA (%) / Sodium perchlorate (mg mL-1) | 0/0 | 0/0 | 0/0 | 0/0 |
| 0.05/6 | 0.05/6 | 0.05/6 | 0.05/6 | |
| 0.1/12 | 0.1/12 | 0.1/12 | 0.1/12 | |
| 0.15/18 | 0.15/18 | 0.15/18 | 0.15/18 | |
Table 1: Tested mobile phase composition composed of water, acetonitrile, trifluoroacetic acid and sodium perchlorate.
Effects of the water percentage on peak resolution
The dependence of the resolution between the peaks corresponding to GSH, GSSG and GSNO on the composition of the mobile phase was evaluated by both partial least squares regression and multiple linear regression analysis. Subsequently, it was possible to determine the optimum mobile phase that provided baseline separated peaks (that is R > 1.5) for all three species in the shortest analysis time (Figure 1). The mobile phase containing higher water/AcN ratios in the mobile phase resulted in considerably longer analysis times and was thus discarded.

Figure 1: Effect of the mobile phase composition on the chromatographic resolution of reduced, oxidised and nitroso-glutathione forms. Numbers indicated on the x axis correspond to the composition of eluent presented in the Table 1. In all cases, RSD values were below 5% (n=4).
Effects of the addition of trifluoroacetic acid to the mobile phase
The addition of a defined amount of TFA to the mobile phase ensured complete protonation of the molecules under investigation and thus symmetric peak shape. An increase in this amount resulted in a poorer reproducibility of the results which was probably due to a poor performance of the chromatographic phase under such acidic conditions. Other acids (e.g. phosphoric or sulphuric) were also evaluated, and showed a similar performance. However, given that TFA was already tested during cell lysis, it was considered as the optimum acid in order to avoid introducing an additional variation into the method that could cause interferences with the sample matrix.
Effects of the sodium perchlorate on GSSG stability
Degradation of GSSG was observed to be negligible during the chromatographic run if at least 12 mg mL-1 of sodium perchlorate were added into the mobile phase containing water/acetonitrile (95/5, v/v) and 0.1% trifluoroacetic acid (Figure 2).

Figure 2: Percentage degradation of GSSG calculated based on sodium perchlorate amount added to the mobile phase (n=3).
Column effectiveness
Column selection was based on the solubility and structure of the group of analytes tested (Table 2). The degree of separation achieved with the selected C18 column was also investigated with two other types of reversed phase C18 columns in order to select the most suitable column for the separation of the three glutathione species.
| Name | Structure | Acidic dissociation constant | Log P | |
|---|---|---|---|---|
| pKa (most acidic) | pKa (most basic) | |||
| GSH | 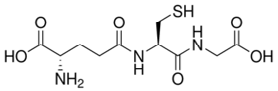 |
2.12 | 9.65 | -3.6 |
| GSSG | 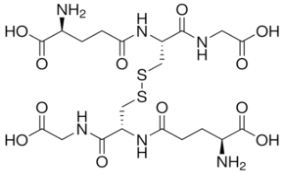 |
2 | 9.61 | -4.9 |
| GSNO | 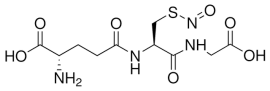 |
2.21 | 9.28 | -2.97 |
Table 2: Chemical characteristics of glutathione molecules.
Columns evaluation was conducted with the aim to identify the best column with which a good separation between selected molecules (resolution), a short time eluting peaks and a good peak shape can be achieved. For this test analysis, columns were selected on the basis of a slight modification in the stationary phase; exploiting the more polar oriented stationary phase, such as the Kinetex, Synergy Fusion column resulted in a confirmation that no overlap with other peaks took place during the optimal separation with the C18 YMC ODS-A column.
Chromatogram runs obtained for these purposes are reported in Figure 3.

Figure 3: Separation of the three glutathione forms on the above mentioned columns using glutathione standards: 1. Reduced glutathione; 2. Oxidized glutathione; 3. S-nitroso-glutathione.
As presented in the zoom of the figure above, a shoulder peak corresponding to the GSSG compound is resulting in the chromatogram when Kinetex column was used. Therefore, the YMC ODS-A column was selected as optimum since better peaks shapes were obtained, no overlapping of glutathione species with matrix components was observed and a shortest analysis time was achieved within 6 minutes. A representative chromatogram of the selected glutathione species determined in A549 cells exposed to NO2 is shown in Figure 4.

Figure 4: Chromatograms (overlay) obtained under optimum conditions with the YMC ODS-A column representing a mixture of GSH, GSSG and GSNO standards and the intracellular glutathione species determined in A549 exposed to NO2.
Optimization of the extraction procedure of intracellular glutathione from A549 cells
As previously published, metal chelators are able to stop enzymatic activities and to prevent the oxidation process of reduced glutathione [39-41]. Therefore, in order to solve both deproteinization and to prevent oxidation of glutathione, effects of various acids were tested for the extraction of intracellular GSH, GSSG and GSNO from the A549 cell line. This was based on the use of low temperatures and the following four lysis buffers: (1) Lysis 1: Cells were incubated at room temperature with 300 μL of MPER lysis buffer; (2) Lysis 2: Cells were incubated at -18°C with 300 μL of a 5% trifluoroacetic acid aqueous solution and scrapped; (3) Lysis 3: Cells were incubated at -18°C with 300 μL of a 5% metaphosphoric acid aqueous solution and scrapped; 4) Lysis 4: Cells were incubated at -18°C with 300 μL of a 0.5% aqueous picric acid solution.
On the other hand, it has been demonstrated that in acidic conditions, artifactual formation of GSNO may appear due to the chemical interaction between GSH and nitrite. As demonstrated by Yap et al., treatment of the biological samples with ammonium sulfamate or N-ethylmaleimide resulted in an accurate assessment of GSNO content [42]. Therefore, in order to accurate determine the GSNO content in the cells, nitrite neutralisation under acidic conditions was considered in our work by adding ammonium sulfate to the acidic solution (0.5% picric acid) prepared to obtain the cellular extract.
The extraction procedure was further carried out according to the steps described in the materials and methods section.
Table 3 shows the amount of GSH extracted from the A549 cells by using each of the above described procedures for the lysis of the cells. As can be observed, the lysate providing the highest GSH concentration is that of picric acid, which was thus selected for further analysis of intracellular GSH, GSSG and GSNO in cells exposed to different chemicals.
| Lysis procedure | GSH [µM] |
|---|---|
| MPER lysis buffer, room temperature | 2.8 |
| 5% TFA, -18°C | 1.7 |
| 5% MPA, -18°C | 0.9 |
| 0.5% picric acid, -18°C | 10.3 |
Table 3: Amount of extracted GSH by each of the lysis procedures. RSD values were below 15% (N=3).
By using picric acid for cell lysis, the detection limit of intracellular glutathiones in the reduced, oxidized and nitroso- forms was in the order of 5 × 104 cells, which is significantly lower than the number of cells required in commercial kits for glutathione analysis (1 × 106 cells).
Analytical performance and method validation
Linearity: Linear calibration curves (8 points for each compound) resulted in straight lines with the following regression equations y=796.62x+15.895, (r2=0.997) for GSH, y=2264.5x+24.081, (r2=0.996) for GSSG and y=2630.4x+18.291, (r2=0.997) for GSNO (Figure 5).

Figure 5: Calibration curves of the three glutathione species (reduced - GSH, oxidised - GSSG, and nitroso-GSNO) where concentrations were plotted against the area units of the peaks within HPLC-UV chromatogram (n=3 ± SD).
Limits of detection and quantification: With the given mobile phase, the LOD and LOQs for GSH, GSSG and GSNO (standard solutions) were calculated based on the calibration curves generated for each individual analyte (Table 4).
| Detection limit [µM] | Quantification limit [µM] | Sensitivity [µM] | |
|---|---|---|---|
| GSH | 0.16 | 0.54 | 0.02 |
| GSSG | 0.08 | 0.26 | 0.01 |
| GSNO | 0.02 | 0.05 | 0.002 |
Table 4: Detection, quantification limits and sensitivity obtained for GSH, GSSG, GSNO within the proposed method.
Accuracy and precision: Glutathione recovery was determined by spiking 6 biological replicates of A549 cells with two different concentrations (3 and 50 μg mL-1) of the standard solutions (GSH, GSSG and GSNO). Following the extraction procedure mentioned above, the cellular extracts were injected immediately in HPLC. The percentage recovery of the present study ranged from 98.7% to 100.1%.
The RSD values for GSH, GSSG and GSNO obtained in the repeatability study ranged from 0.05 to 0.16%; 0.01 to 0.6% and 0.02 to 0.09%. Based on the evaluation of the long term reproducibility of the method the RSD obtained was <5% for all three glutathione species (0.9-2.7% for GSH; 0.4-2.9% for GSSG and 0.2-1.4% for GSNO).
Storage of the standard solutions and of the cellular extracts: Standard solutions stability during the analysis was checked at the beginning and the end of analysis. Additionally, the potential degradation of standards stored under different conditions (4, -20, -80°C) was verified. These temperatures were selected so that any enzymatic activity in the cells is discontinued, thus minimizing the risk of any species inter-conversion, from the moment when cell exposure to the target pollutants finishes and the analysis of intracellular glutathione species in the cells takes place.
Standards stored at 4°C showed to be stable for up to 3 days (7% RSD), and for up to 4 months at -80°C (5% RSD).
Applicability of the glutathione method on A549 cell line exposed to various chemicals
Effects of R-limonene, limonene oxide and nitric dioxide on cell viability: Both R-limonene and limonene oxide did not significantly alter the cell viability after up to 24 hours according to experiments carried out by NRU. On the other hand, exposing the cells to nitric dioxide at air-liquid interface for 1 and 2 hours, showed a statistically significant decrease in cellular viability after overnight post-incubation (data not shown). Therefore, the modification in the intracellular glutathione content by NO2 was analysed immediately after cells exposure, in order to avoid underestimation of the glutathione amount due to the loss of cell viability that was observed after overnight postincubation.
GSH, GSSG and GSNO quantification in human lung cells: Under basal conditions, the GSH content that was measured in A549 cells by our HPLC-UV method gives values of 34.75 ± 0.3 nmole mg-1 protein, n=8, which are in agreement with those reported by A. Spadaro et al. (e.g. 30.11 ± 1.53 nmole mg-1 protein, n=6) [9].
Effects of R-limonene and limonene oxide on glutathione content in A549 cells (test chemicals solubilized into the culture media):The optimised HPLC method (as described above) was applied for the analysis of biological samples, A549 cells - a common cell lines used in cancer research - exposed to different inhalable chemicals.
The results obtained showed that the GSH:GSSG ratio slightly increased to 1.4-fold (n=3, p<0.01) and GSH:GSNO ratio increased to 1.5-fold, in the case of cells exposed to limonene oxide (137.2 mg L-1) when compared to both GSH:GSSG and GSH:GSNO ratios of untreated cells. These results might suggest that, limonene oxide stimulates a de novo synthesis of GSH, since no significant increase was observed in the amount of oxidized or nitroso- glutathione forms, but only in the amount of reduced glutathione form. On the other hand, cells exposed to R-limonene, did not present any significant variation in any of the three glutathione species compared to the cells untreated.
Effects of nitric dioxide on glutathione content in A549 cells (air-liquid interface culture conditions): As presented in Figure 6, the GSH:GSSG ratio decreased 2.3 times, while the GSH:GSNO ratio significantly decreased to 9.5 times (p<0.01) in A549 cells exposed for 1 hour to NO2 (12 ppm) when compared to untreated cells. After 2 hours of exposure, the GSH levels decreased 1.7-fold compared to the negative control, and 1.3-fold compared to the GSH amount obtained after 1 hour. A significant increase in GSNO was observed after 2 hours, corresponding to 3 times higher than the level found in cells untreated suggesting that glutathione played an active role in the detoxification of NO2 in the cells.

Figure 6: Intracellular % of GSH: GSSG and GSH: GSNO of cells exposed in triplicate to NO2 for 1 hour). (n=3, average ± 1SD, **p<0.01).
The concomitant determination of the three glutathione species in human cells facilitates studies aiming at better understanding glutathione’s detoxification abilities/properties towards reactive oxygen and nitrogen species.
In accordance with previous studies, no significant effect on the glutathione content was observed when human lung fibroblasts were exposed for 24 hrs to R-limonene (up to 200 μM) [43]. Intracellular levels of GSH and GSSG may be decreased or not affected by some oxides (e.g. ethylene oxide) while others may induce increased activities of some forms of intracellular glutathione (e.g. nitric oxide) [44-47]. Since no significant increase was observed for oxidized gluthathione, the authors concluded that the most probable detoxification route seems to be a complexation between the thiol group in reduced glutathione and oxides [48]. Further research is however needed in order to better understand how epoxides can be detoxified in the cell. Considering its oxidant potential, and that previous works have pointed towards the variation of GSH content in mice exposed to NO2, in the present work it was evaluated whether NO2 is able to alter the intracellular GSH level in human pulmonary cells. This may then allow us to study the mechanism involved in such interactions, by measuring both oxidized GSH and S-nitroso-GSH. To do so, both the cell lysis procedure and the HPLC-UV analytical method for the intracellular glutathione forms, were optimized focusing on GSH, GSSG and GSNO.
Conclusion
A key objective of the present study was to develop and optimise an analytical method based on liquid chromatography coupled with a UV detector to simultaneously identify and quantify GSH, GSSG and GSNO in human epithelial cells.
In a recent paper, the three glutathione forms GSH, GSSG, GSNO were simultaneously identified in plant species (e.g. pepper plant organs) by using a liquid chromatography-electrospray/mass spectrometry method (LC-ES/MS) [48]. Based on our outcomes, the HPLC-UV method appears to be faster, in terms of both time of analysis and time required for sample preparation, in comparison with the LC-ES/MS method applied to quantify the glutathione content in plants.
In this sense, a fast and reproducible method, based on liquid chromatography, coupled to UV detection, was developed and optimised in order to identify and quantify free glutathione species (GSH, GSSG, and GSNO) produced in human cells. Therefore, in order to validate this HPLC-UV method; parameters such as limit of detection, limit of quantification, reproducibility and repeatability were evaluated.
The A549 cell line which we exposed to various chemicals, by two in vitro exposure methods (direct addition of chemicals into culture media and chemicals applied to air-lifted cells), demonstrates the applicability of the method for the quantification of the three intracellular glutathione species found in human pulmonary cells. The GSH basal values that we determined in our cell line are comparable to a recent study in which samples were analysed by HPLC with electrochemical detection [9]. In addition to the studied A549 cells, the method presented here can be easily applied to the quantification of GSH, GSSG and GSNO in a variety of other human cell lines (e.g. the applicability of the HPLCUV method for the quantification of glutathione species reduced-GSH, oxidised-GSSG, nitroso-GSNO was also carried out for the human bronchial epithelial cell line 16HBE14o-, data not shown here) due to the short sample preparation, no need for derivatisation procedure, those being important steps to minimize the risk of underestimation or overestimation of glutathione content. Therefore, the proposed method shows an alternative way to determine, in a relatively short length of time, each of the above-mentioned glutathione forms in human cell cultures.
Acknowledgements
The authors would hereby like to gratefully acknowledge the financial support from OFFICAIR project (On the reduction of health effects from combined exposure to indoor air pollutants in modern offices, funded by the European Union 7th Framework (Agreement 265267) under the Theme: ENV.2010.1.2.2-1) and to extend their gratitude to Mr. Bo Larsen, Mr. Dimitris Kotzias and Mr. Otmar Geiss for useful discussions and suggestions. We would also like to acknowledge the contribution of Ms. Margaret Holland in proof-reading and correcting the paper.
References
- Lushchak VI (2012) Glutathione homeostasis and functions: potential targets for medical interventions. J Amino Acids 2012: 736837.
- Sies H (1999) Glutathione and its role in cellular functions. Free Radic Biol Med 27: 916-921.
- Pastore A, Federici G, Bertini E, Piemonte F (2003) Analysis of glutathione: implication in redox and detoxification. Clin Chim Acta 333: 19-39.
- Jozefczak M, Remans T, Vangronsveld J, Cuypers A (2012) Glutathione is a key player in metal-induced oxidative stress defenses. Int J Mol Sci 13: 3145-3175.
- Carmel-Harel O, Storz G (2000) Roles of the glutathione- and thioredoxin-dependent reduction systems in the Escherichia coli and saccharomyces cerevisiae responses to oxidative stress. Annu Rev Microbiol 54: 439-461.
- Wu G, Fang YZ, Yang S, Lupton JR, Turner ND (2004) Glutathione metabolism and its implications for health. J Nutr 134: 489-492.
- Lipton AJ, Johnson MA, Macdonald T, Lieberman MW, Gozal D, et al. (2001) S-nitrosothiols signal the ventilatory response to hypoxia. Nature 413: 171-174.
- Martín MA, Ramos S, Mateos R, Granado Serrano AB, Izquierdo-Pulido M, et al. (2008) Protection of human HepG2 cells against oxidative stress by cocoa phenolic extract. J Agric Food Chem 56: 7765-7772.
- Spadaro A, Ronsisvalle G, Pappalardo M (2011) Rapid Analysis of Glutathione in Human Prostate Cancer Cells (DU145) and Human Lung Adenocarcinoma Cells (A549) by HPLC with Electrochemical Detection. J Pharm Sci Res 3: 1637-1641.
- Anderson ME (1998) Glutathione: an overview of biosynthesis and modulation. Chem Biol Interact 111-112: 1-14.
- Hassan HM, Fridovich I (1980) Mechanism of the antibiotic action pyocyanine. J Bacteriol 141: 156-163.
- Liu H, Wang H, Shenvi S, Hagen TM, Liu RM (2004) Glutathione metabolism during aging and in Alzheimer disease. Ann N Y Acad Sci 1019: 346-349.
- Liu H, Harrell LE, Shenvi S, Hagen T, Liu RM (2005) Gender differences in glutathione metabolism in Alzheimer's disease. J Neurosci Res 79: 861-867.
- Cai J, Chen Y, Seth S, Furukawa S, Compans RW, et al. (2003) Inhibition of influenza infection by glutathione. Free Radic Biol Med 34: 928-936.
- Ichinose S, Nakamura M, Maeda M, Ikeda R, Wada M, et al. (2009) A validated HPLC-fluorescence method with a semi-micro column for routine determination of homocysteine, cysteine and cysteamine, and the relation between the thiol derivatives in normal human plasma. Biomed Chromatogr 23: 935-939.
- Chwatko G, Kuzniak E, Kubalczyk P, Borowczyk K, Wyszczelska-Rokiel M, et al. (2014) Determination of cysteine and glutathione in cucumber leaves by HPLC with UV detection. Anal Methods 6: 8039-8044.
- Michaelsen JT, Dehnert S, Giustarini D, Beckmann B, Tsikas D (2009) HPLC analysis of human erythrocytic glutathione forms using OPA and N-acetyl-cysteine ethyl ester: evidence for nitrite-induced GSH oxidation to GSSG. J Chromatogr B Analyt Technol Biomed Life Sci 877: 3405-3417.
- Tsikas D, Sandmann J, Holzberg D, Pantazis P, Raida M, et al. (1999) Determination of S-nitrosoglutathione in human and rat plasma by high-performance liquid chromatography with fluorescence and ultraviolet absorbance detection after precolumn derivatization with o-phthalaldehyde. Anal Biochem 273: 32-40.
- Baker MA, Cerniglia GJ, Zaman A (1990) Microtiter plate assay for the measurement of glutathione and glutathione disulfide in large numbers of biological samples. Anal Biochem 190: 360-365.
- Senft AP, Dalton TP, Shertzer HG (2000) Determining glutathione and glutathione disulfide using the fluorescence probe o-phthalaldehyde. Anal Biochem 280: 80-86.
- Blanco RA, Ziegler TR, Carlson BA, Cheng PY, Park Y, et al. (2007) Diurnal variation in glutathione and cysteine redox states in human plasma. Am J Clin Nutr 86: 1016-1023.
- Guvenc M, Cetintas B, Irtegun S, Tastan H, Sahin K (2009) A Practical HPLC Method to Measure Reduced (GSH) and Oxidized (GSSG) Glutathione Concentrations in Animal Tissues. Journal of Animal and Veterinary Advances 8: 343-347.
- Gan J, Harper TW, Hsueh MM, Qu Q, Humphreys WG (2005) Dansyl glutathione as a trapping agent for the quantitative estimation and identification of reactive metabolites. Chem Res Toxicol 18: 896-903.
- Serru V, Baudin B, Ziegler F, David JP, Cals MJ, et al. (2001) Quantification of reduced and oxidized glutathione in whole blood samples by capillary electrophoresis. Clin Chem 47: 1321-1324.
- Afzal M, Afzal A, Jones A, Armstrong D (2002) A rapid method for the quantification of GSH and GSSG in biological samples. Methods Mol Biol 186: 117-122.
- Steffen M, Sarkela TM, Gybina AA, Steele TW, Trasseth NJ, et al. (2001) Metabolism of S-nitrosoglutathione in intact mitochondria. Biochem J 356: 395-402.
- World Health Organization (2007) Global surveillance, prevention and control of chronic respiratory diseases: A comprehensive approach.
- Adgate JL, Church TR, Ryan AD, Ramachandran G, Fredrickson AL, et al. (2004) Outdoor, indoor, and personal exposure to VOCs in children. Environ Health Perspect 112: 1386-1392.
- Wallace LA (1983) Volatile organic compounds. In: Indoor air pollution: a health perspective. Johns Hopkins University Press 199: 252-272.
- Marine SS, Clemons J (2003) Determination of limonene oxidation products using SPME and GC-MS. J Chromatogr Sci 41: 31-35.
- Koistinen K, Kotzias D, Kephalopoulos S, Schlitt C, Carrer P, et al. (2008) The INDEX project: executive summary of a European Union project on indoor air pollutants. Allergy 63: 810-819.
- World Health Organization (2006) Air quality guidelines: global update: Particulate matter, ozone, nitrogen dioxide and sulfur dioxide. WHO Regional Office for Europe.
- HSDB: Picric acid. In: Hazardous Substances Data Bank”, National Library of Medicine, Bethesda, MD, USA.
- Broniowska KA, Diers AR, Hogg N (2013) S-nitrosoglutathione. Biochim Biophys Acta 1830: 3173-3181.
- Hogg N (2002) The biochemistry and physiology of S-nitrosothiols. Annu Rev Pharmacol Toxicol 42: 585-600.
- Lipsa D, Cacho C, Rembges D, Barrero-Moreno J (2014) Fast cell counting - the better cell counting? Fresenius Environmental Bulletin 23: 3054-3058.
- Pariselli F, Sacco MG, Rembges D (2009) An optimized method for in vitro exposure of human derived lung cells to volatile chemicals. Exp Toxicol Pathol 6: 33-39.
- Repetto G, del Peso A, Zurita JL (2008) Neutral red uptake assay for the estimation of cell viability/cytotoxicity. Nat Protoc 3: 1125-1131.
- Rebrin I, Forster MJ, Sohal RS (2007) Effects of age and caloric intake on glutathione redox state in different brain regions of C57BL/6 and DBA/2 mice. Brain Res 1127: 10-18.
- Han D, Hanawa N, Saberi B, Kaplowitz N (2006) Hydrogen peroxide and redox modulation sensitize primary mouse hepatocytes to TNF-induced apoptosis. Free Radic Biol Med 41: 627-639.
- Rossi R, Milzani A, Dalle-Donne I, Giustarini D, Lusini L, et al. (2002) Blood glutathione disulfide: in vivo factor or in vitro artifact? Clin Chem 48: 742-753.
- Yap LP, Sancheti H, Ybanez MD, Garcia J, Cadenas E, et al. (2010) Determination of GSH, GSSG, and GSNO using HPLC with electrochemical detection. Methods Enzymol 473: 137-147.
- Rolseth V, Djurhuus R, Svardal AM (2002) Additive toxicity of limonene and 50% oxygen and the role of glutathione in detoxification in human lung cells. Toxicology 170: 75-88.
- Katoh T, Higashi K, Inoue N, Tanaka I (1989) Lipid peroxidation and the metabolism of glutathione in rat liver and brain following ethylene oxide inhalation. Toxicology 58: 1-9.
- Fujishiro K, Mori K, Inoue N (1991) Effects of inhaled ethylene oxide on the lens glutathione redox cycle in rats. Arch Toxicol 65: 606-607.
- Calabrese V, Scapagnini G, Ravagna A, Bella R, Foresti R, et al. (2002) Nitric oxide synthase is present in the cerebrospinal fluid of patients with active multiple sclerosis and is associated with increases in cerebrospinal fluid protein nitrotyrosine and S-nitrosothiols and with changes in glutathione levels. J Neurosci Res 70: 580-587.
- Sundberg K, Dreij K, Seidel A, Jernström B (2002) Glutathione conjugation and DNA adduct formation of dibenzo[a,l]pyrene and benzo[a]pyrene diol epoxides in V79 cells stably expressing different human glutathione transferases. Chem Res Toxicol 15: 170-179.
- Airaki M, Sánchez-Moreno L, Leterrier M, Barroso JB, Palma JM, et al. (2011) Detection and quantification of S-nitrosoglutathione (GSNO) in pepper (Capsicum annuum L.) plant organs by LC-ES/MS. Plant Cell Physiol 52: 2006-2015.
Relevant Topics
Recommended Journals
Article Tools
Article Usage
- Total views: 18355
- [From(publication date):
August-2015 - Dec 22, 2024] - Breakdown by view type
- HTML page views : 13607
- PDF downloads : 4748