Research Article Open Access
Determination of Intramolecular 13C Isotope Distribution of Pyruvate by Headspace Solid Phase Microextraction-Gas Chromatography-Pyrolysis-Gas Chromatography-Combustion- Isotope Ratio Mass Spectrometry (HS-SPMEGC-Py-GC-C-IRMS) Method
Tarin Nimmanwudipong1, Naizhong Zhang3, Alexis Gilbert2, Keita Yamada3 and Naohiro Yoshida1,2,3
1Department of Environmental Science and Technology, Tokyo Institute of Technology, 4259 Nagatsuta, Midori-ku, Yokohama 226-8502, Japan
2Earth-Life Science Institute (WPI-ELSI), Tokyo Institute of Technology, Meguro, Tokyo 152-8551, Japan
3Department of Environmental Chemistry and Engineering, Tokyo Institute of Technology, 4259 Nagatsuta, Midori-ku, Yokohama 226-8502, Japan
- *Corresponding Author:
- Tarin Nimmanwudipong
Department of Environmental Science and Technology
Tokyo Institute of Technology, 4259 Nagatsuta
Midoriku, Yokohama 226-8502, Japan
Tel: +81459245517
Fax: +81459245517
E-mail: Nimmanwudipong.t.aa@m.titech.ac.jp
Received Date: Novmber 27, 2015; Accepted Date: December 21, 2015; Published Date: December 28, 2015
Citation: Nimmanwudipong T, Zhang N, Gilbert A, Yamada K, Yoshida N (2015) Determination of Intramolecular 13C Isotope Distribution of Pyruvate by Headspace Solid Phase Microextraction-Gas Chromatography-Pyrolysis-Gas Chromatography-Combustion- Isotope Ratio Mass Spectrometry (HS-SPMEGC-Py-GC-C-IRMS) Method. J Anal Bioanal Tech 7:293. doi:10.4172/2155-9872.1000293
Copyright: © 2015 Nimmanwudipong T, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Visit for more related articles at Journal of Analytical & Bioanalytical Techniques
Abstract
This paper presents the improvement of analytical methods for intramolecular carbon isotope compositions (δ13C) of pyruvate. Decarboxylated by H2O2, pyruvate yields acetic acid and CO2. Headspace solid phase micro-extractiongas chromatography-pyrolysis-gas chromatography-combustion-isotope ratio mass spectrometry (HS-SPME-GCPy- GC-C-IRMS) was used to measure the intramolecular δ13C values of acetic acid. δ13C value of CO2 can be later calculated using mass balance equation. The method’s consistency was confirmed by comparison of the δ13C value of CO2 from calculation to its direct measurement. Results of this study confirmed the method improvement because pyruvate 13C intramolecular distribution patterns were obtained. Two intramolecular 13C distribution patterns for commercial chemical reagents were found using this developed method. Intramolecular 13C distribution patterns for pyruvate were found for application in dietary supplements. Its origin was inferred. The method presented herein is expected to be a useful tool for categorization of pyruvate into different intramolecular 13C distribution patterns, which might indicate different production processes or raw materials.
Keywords
Pyruvate; Dietary supplement; Intramolecular isotope distribution; Food authenticity; Quality control
Introduction
Isotope analysis has been applied to facilitate identification of the origin, metabolic pathways, and biosphere-atmosphere interaction of organic materials [1-4]. Compound Specific Isotope Analysis (CSIA) is typically used to ascertain the isotopic composition of a target compound. However, in some cases, CSIA data alone are not sufficient for identification of the compound’s origin. Position-Specific Isotope Analysis (PSIA) has provided information related to the heterogeneous isotope distribution of organic compounds including amino acids, acetic acid, fatty acids, sugars, ethanol, and hydrocarbons [5-9]. This information is crucially important for the investigation of synthetic processes and metabolic pathways of the target compound. These isotope analysis techniques are also applied in the food industry for quality control [1,2].
Pyruvate, a key metabolite for carbohydrate metabolism, is necessary to trigger a plant’s citric acid cycle, fat, and protein metabolism. It can also be useful as a dietary supplement to increase the metabolic rate [10]. Pyruvate influences the isotopic contents of respired CO2 and its related metabolites. Therefore, its isotope signature would be beneficial for studying the authenticity and metabolic pathways in plants.
The main objective of this study is to improve the analytical method for intramolecular 13C distribution of pyruvate, which can be degraded into acetic acid and CO2 using H2O2 [11,12]. Results confirm the success of this technique: we obtained a pyruvate 13C intramolecular distribution pattern.
Additionally, we applied this method to ascertain the intramolecular 13C distribution in a pyruvate sample from pyruvate supplement pills. Dietary supplements are convenient choices that provide essential nutrients. Because of their high demand, the number of manufacturers has increased rapidly in the past few years. One way to remain competitive in the market is to decrease manufacturing costs to the greatest extent possible. Commonly, people prefer authentic products from natural sources. However, the same commercial synthetic substance, which is obtainable by the derivatization of petroleum or coal, offers a rapidly producible and cheaper alternative than the natural extract from biogenic sources [13-15]. According to the Food and Drug Administration (FDA) in some countries, synthetic substances are illegal: their manufacture is prohibited [16]. To minimize the risk of illegality, a method is needed to help differentiate between synthetic and natural substances [17-19]. This study considered the potential origin of pyruvate in dietary supplement based on the intramolecular 13C distribution.
Materials and Methods
Annotations
The carbon isotope composition in per mil (‰) concentrations is expressed as the δ13C value, the carbon isotope ratio (13C/12C) of the sample against an international standard (VPDB).
 (1)
(1)
For this study, pyruvate samples were measured after decarboxylation, which yields acetic acid and carbon dioxide. Bulk and intramolecular δ13C values of pyruvate are definable in a mass balance equation as
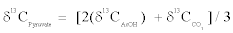 (2)
(2)
Therein, δ13CAcOH value is the bulk carbon isotope composition of acetic acid and δ13CCO2 value is the carbon isotope composition of CO2 from pyruvate decarboxylation in this study. Actually, bulk and intramolecular δ13C values of acetic acid are definable in a mass balance equation as
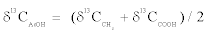 (3)
(3)
Where δ13CCH3 value and δ13CCOOH value respectively represents the carbon isotope composition of methyl and carboxyl carbon atom of acetic acid. Each carbon position of pyruvate was designated as C-1 (carboxyl part), C-2 (carbonyl part), and C-3 (methyl part).
Chemicals
Four sodium pyruvates designated as A (Tokyo Chemical Industry Co. Ltd., Tokyo, Japan), B (MP Biomedicals, LLC, CA, USA), C (Sigma- Aldrich Corp., St. Louis, MO, USA), and D (Wako Pure Chemical Industries Ltd., Osaka, Japan) were used for this study. Four pyruvate supplement samples, designated as DP1 (Earth Natural Supplements, Florida, USA), DP2 (Best Naturals, NJ, USA), DP3 (Source Naturals, Inc., CA, USA) in capsule pills, and DP4 (Now Foods, IL, USA) in tablet pills were purchased. Hydrogen peroxide (30%) and hydrochloric acid (0.1 mM) (Wako Pure Chemical Industries Ltd.) were used respectively for pyruvate decarboxylation and pH adjustment. A tin capsule (0.15 ml: ∅ 5/19 mm; LÜDI Swiss AG, Switzerland) was used to contain sodium pyruvate samples for laser spectroscopy analysis.
Degradation of pyruvate
H2O2-catalyzed decarboxylation of pyruvate is described according to the following scheme:
CH3COCOOH+H2O2 → CH3COOH+CO2+H2O (4)
In this study, sodium pyruvate samples (A, B, C, and D) were degraded using 30% hydrogen peroxide, then yielding acetic acid, CO2 and H2O as products, as described below. The yields of acetic acid at 10, 30, 60 and 120 min of degradation time were determined using ion chromatography (IC-20 Dionex™; Thermo Fisher Scientific Inc., Bremen, Germany).
For the degradation of sodium pyruvate, a pyruvate aqueous solution was prepared at 85 mM diluting with distilled water. For pyruvate supplement samples, pyruvate was separated from other ingredients before dilution with distilled water. One pill of 750 mg (DP2, DP3) and 1000 mg (DP1, DP4) was used for pyruvate extraction. Regarding to packages’ label, pyruvate supplementary samples have main ingredients consists of pyruvate salt, gelatin (contained capsule), stearate, and cellulose. The gelatin container was taken off and discarded (DP1, DP2, DP3); the powder sample was kept. The DP4 tablet was crushed to powder in ceramic mortar. Remaining powder of samples was diluted in 100 mL milliQ water. Because of the lack of water solubility, stearate and cellulose were separated using microfiltration three times using a 20 μm filter. Then the pyruvate aqueous solution was obtained from supplement samples.
In a 20 mL gas-tight vial, 1 mL of each pyruvate aqueous solution sample was put in and topped with rubber cap for analysis. To samples for pyruvate degradation, 0.2 mL of H2O2 was added. After complete degradation, the samples’ pH was adjusted to 1.0-2.0 pH by adding 0.2 mL of 0.1 mol/L HCl. The CO2 derived from the degradation was collected and purified by repeated cryogenic method and trapped in the Pyrex® sealed tube for δ13C analysis.
Carbon isotopic analysis
Intramolecular δ13C value of acetic acid derived from pyruvate degradation was measured using HS-SPME-GC-Py-GC-C-IRMS [20]. The system consists of a first gas chromatograph, (TraceTM GC Ultra; Thermo Fisher Scientific Inc.) equipped with a capillary column (NukolTM, 30 m × 0.32 mm i.d., 1 μm film thickness; Supelco, PA, USA), connected to a second gas chromatograph (HP 6890 series; Hewlett-Packard Co., PA, USA) equipped with a second capillary column (HP-Plot Q 30 m × 0.32 mm i.d., 20 μm film thickness; Agilent Technologies Inc., CA, USA). Two gas chromatographs were connected through a pyrolysis furnace part (ceramic tube, 25 cm × 0.5 mm i.d.) operated at 1000°C for pyrolysis of acetic acid. The pyrolytic products were separated using a second capillary column and were introduced into a combustion furnace (ceramic tube, 25 cm × 0.5 mm i.d., packed with CuO, NiO, and Pt wires) operated at 960°C. The second chromatograph was connected via Thermo GC Isolink™ and Conflo-IV™ interfaces (both from Thermo Fisher Scientific Inc.) to a mass spectrometer (Finnigan Delta V™; Thermo Fisher Scientific Inc.). A transfer line between chromatographs was made using deactivated fused silica capillary (0.32 mm i.d.; GL Sciences Inc., Japan).
Acetic acid from pyruvate degradation was extracted using an SPME device, equipped with 85 μm SPME fiber coated with carboxen/ polydimethylsiloxane (Carboxen/PDMS stableflex™; Supelco, PA, USA). Extraction was conducted in a thermostatic chamber controlled to 25°C: the non-stirred samples condition. The extraction time was 60 min. After HS-SPME extraction, the fiber was inserted into the injection port of the first gas chromatograph at 250°C. Helium was used as a carrier gas for all experiments. Chromatographic conditions were the following: 2.0 mL/min flow rate of carrier gas and 10:1 split ratio. The first oven temperature program was the following: 100°C (5 min), then rising to 190°C (10 min) at the rate of 15°C/min, and finally at 200°C (2 min) at the rate of 15°C/min. The second gas chromatograph was kept constantly at 40°C.
The dual-inlet system of an isotope ratio mass spectrometer (MAT 253TM, Thermo Fisher Scientific Inc.) was used for the measurement of δ13C value of CO2 derived from pyruvate degradation. Bulk δ13C values of sodium pyruvate (A, B, C, and D) were measured using cavity ringdown laser spectroscopy (Picarro G1121i; Picarro Inc., CA, USA).
Results and Discussion
Completeness of reaction and consistency of method
The experiment of pyruvate decarboxylation by H2O2 was conducted respectively in ranges of 10, 30, 60 and 120 min. The acetic acid yield was measured using ion chromatography and was calculated using a calibration curve of the acetic acid standard. As shown in Table 1, the yield of acetic acid reaches 99% at 60 min reaction time. At the 120 min range, it also had the same number around 99%, which implies that the reaction is completed at 60 min. For subsequent experiments, we used 60 min as the decarboxylation time.
| Degradation time (min.) | Yield of acetic acid (%) |
|---|---|
| 10 | 76.4 |
| 30 | 86.2 |
| 60 | 99.1 |
| 120 | 99.9 |
Table 1: Yield percentage of acetic acid by degradation time.
The consistency of δ13CC-1 (δ13C value of C-1 of pyruvate) was confirmed by comparison of δ13CCO2 between the value calculated using the mass balance equation of pyruvate (equation 2) and from direct measurement, which are expected to be the same. Table 2 shows that the differences of δ13CCO2 values from the two methods were approximately 0.6‰, which is an acceptable range, showing that usage of the mass balance equation can obtain δ13CCO2 value (equation 2). This consistency of method has also confirmed the acceptable use of HSSPME- GC-Py-GC-C-IRMS, for which δ13C of C-2 and C-3 (δ13C value of C-2 and C-3 of pyruvate) are obtainable in a single step. Without the measurement of bulk δ13CAcOH value, the δ13C measurement can reduce the unexpected errors occurs by duplicate sample preparation or switching between configuration systems [20]. δ13CC-1 value can be calculated later using the mass balance equation of pyruvate.
| Sample (n=3) | Calculation (‰) | Measurement (‰) | Difference (‰) |
|---|---|---|---|
| A | -20.0 ± 0.7 | -19.4 ± 0.1 | 0.6 |
| B | -25.3 ± 0.6 | -24.8 ± 0.3 | 0.5 |
| C | -15.2 ± 0.7 | -15.5 ± 0.0 | 0.3 |
| D | -20.9 ± 0.1 | -20.3 ± 0.3 | 0.6 |
Table 2: Mass balance calculation and direct measurement of δ13CCO2 (C-1).
Bulk and intramolecular δ13C isotope distribution of sodium pyruvate
Details of δ13C values of sodium pyruvate samples are presented in Table 3. First, we obtained bulk δ13C of sodium pyruvate, which are -22.6‰ (A), -22.6‰ (B), -21.3‰ (C), and -23.1‰ (D). For intramolecular δ13C values, samples A, C, and D have δ13C values in the pattern of C-3>C-1>C-2, whereas sample B has the δ13C pattern of C-2>C-3>C-1. Figure 1 clarifies that we obtained intramolecular δ13C distribution of pyruvate of two kinds. Moreover, same bulk δ13C value of A and B samples have different patterns of intramolecular δ13C values. These indicate that these pyruvates are potentially derived from different production processes or raw materials.
| Sample | δ13CC-1 (‰) | δ13CC-2 (‰) | δ13CC-3 (‰) | δ13CAcOH (‰) | Bulk δ13C (‰) |
|---|---|---|---|---|---|
| A | -20.0 ± 0.7a | -36.5 ± 0.3 | -11.3 ± 0.4 | -23.89 ± 0.5 | -22.6 ± 0.2 |
| B | -25.3 ± 0.6 | -19.9 ± 0.2 | -22.7 ± 0.3 | -21.26 ± 104 | -22.6 ± 0.2 |
| C | -15.2 ± 0.7 | -36.6 ± 0.4 | -12.0 ± 0.5 | -24.27 ± 0.6 | -21.3 ± 0.2 |
| D | -20.9 ± 0.1 | -35.6 ± 0.5 | -12.7 ± 0.5 | -24.14 ± 0.7 | -23.1 ± 0.0 |
| DP1 | -36.6 ± 0.8 | -21.2 ± 0.3 | -29.3 ± 0.1 | -25.23 ± 0.3 | -29.0 ± 1.5 |
| DP2 | -34.8 ± 1.5 | -16.5 ± 0.3 | -23.3 ± 0.3 | -19.92 ± 0.4 | -24.9 ± 2.7 |
| DP3 | -39.0 ± 1.5 | -17.6 ± 0.0 | -23.3 ± 0.3 | -20.46 ± 0.3 | -26.7 ± 2.6 |
| DP4 | -34.9 ± 0.3 | -18.4 ± 0.2 | -24.6 ± 0.2 | -21.51 ± 0.3 | -26.0 ± 0.7 |
| Sodium pyruvate using H2O2 degradation [11] | -22.3 | -19.6 | -21.5 | -20.57 | -21.2 |
aStandard deviation from the mean (n=3)
Table 3: Δ13C measurement of sodium pyruvate samples and degraded fragments with Δ13C calculated from the mass balance equation and measurement values of Δ13C.

Figure 1: δ13C pattern of commercial sodium pyruvate (A-D), pyruvate from diet supplement (DP1-DP4) and sodium pyruvate from a previous study [11].
Pyruvate can be synthesized using chemical production, with tartaric acid and KHSO4 as substrates [21]. Tartaric acid has two main pathways for chemical synthesis [22,23]. First is tartaric acid obtained from petroleum by-products, which inherit the δ13C value from hydrocarbon substrate. Second is tartaric acid from cyanohydrin synthesis, which inherits the δ13C value from the initial substrate (3 carbons from glyceraldehyde and 1 carbon from the cyano group). Recently, Zyakun et al. determined the intramolecular δ13C value in synthetic tartaric acid from chemical synthesis, finding 13C depletion in its carboxyl carbon [23]. In general, without isotope fractionation, pyruvate is expected to inherit δ13C value of the beginning tartaric acid. However, isotope fractionation can occur during actual production processes. Our hypothesis according to previous works is tartaric acid from chemical synthesis, which also has a similar trend of δ13C values to acetic acid from chemical synthesis, which has depleted carboxyl carbon [8,23]. For intramolecular δ13C value of pyruvate, the depletion of δ13C value in carboxyl carbon (C-1) from C-2 and C-3 has been found in sample B. This trend is similar to the trend of intramolecular δ13C values of acetic acid [8,24] and tartaric acid [23] from chemical synthesis. We might infer that sample B had high potential to be produced by a chemical synthesis method, along with a good agreement to the δ13C values of previous studies. However, without details of proprietary synthetic process of sample, the discussion about the exact pattern remains unclear. Further details related to isotope fractionation, which possibly occurred in production process, must be clarified in future works’ discussion for concrete references.
Another pattern of intramolecular δ13C values might have a different mode of production or substrate. Pyruvate can also be produced using biotechnological methods. Biotechnological methods have at least three methods: direct fermentation method, the resting cell method, and the enzymatic method [25]. The enzymatic method is simple, with a high conversion rate of the substrate. For example, lactate can be the substrate for pyruvate production using L-lactate catalyzed by glycolate oxidase in Hansenula polymorpha [26]. However, the high price of raw materials and some complicated processes for removal of by-products of production are shortcomings related to industrialize enzymatic methods for pyruvate production. Consequently, direct fermentation and the resting cell methods have higher potential for mass production of pyruvate. Samples A, C, and D have found enrichment in δ13C value in C-1 than C-2 and C-3, which also have the same trend of δ13C values as those of biological products reported from previous studies [8,23]. In this case, samples A, C, and D should have been regarded as products from biotechnological methods. We also considered the pattern of δ13C values of the sodium pyruvate sample that used H2O2 degradation in a previous study [11], which has a similar pattern to that of sample B and which should fall into the category of chemical synthesis production, from the 13C depletion in its carboxyl carbon than its C-2 and C-3.
Considering pyruvate supplement samples, we found their intramolecular 13C distribution patterns to be similar to sample B, which is potentially, produced using chemical synthesis methods. However, if natural tartaric acid is the initial substance in chemical synthesis of pyruvate, then the intramolecular 13C pattern might be different, according to the different pattern of δ13C values of biogenic and abiogenic tartaric acid [23]. These intramolecular 13C distributions of pyruvate can help us categorize the production process of pyruvate, although further investigation of the intramolecular 13C distribution pattern from plenty of natural samples and samples that are different from known processes must be done for additional explanations.
Conclusion
Adoption of HS-SPME-GC-Py-GC-C-IRMS produces a more convenient analytical method for the determination of intramolecular δ13C values in pyruvate. Pyruvate samples in this study have two patterns that are useful for categorizing samples into different production processes. Further studies of the natural pattern of the pyruvate from plants can be a good first step, followed by studies of pyruvate production by different known processes. These will help to distinguish the pyruvate samples into the correct categories of origin processes.
Acknowledgements
This study was supported by a Grant in-Aid for Scientific Research (S) (23224013). A. Gilbert appreciates a Grant-in-Aid for Young Scientists (B) (15K17774), and thanks MEXT, Japan, for financial support.
References
- Rossmann (2001) Determination of stable isotope ratios in food analysis. Food Rev Int 17: 347-381.
- Förstel H (2007) The natural fingerprint of stable isotopes--use of IRMS to test food authenticity. Anal Bioanal Chem 388: 541-544.
- Brenna JT (2001) Natural intramolecular isotope measurements in physiology: elements of the case for an effort toward high-precision position-specific isotope analysis. Rapid Commun Mass Spectrom 15: 1252-1262.
- Gilbert A, Robins RJ, Remaud GS, Tcherkez GG (2012) Intramolecular 13C pattern in hexoses from autotrophic and heterotrophic C3 plant tissues. Proc Natl Acad Sci USA 109: 18204-18209.
- Gilbert A, Silvestre V, Robins RJ, Remaud GS (2009) Accurate quantitative isotopic 13C NMR spectroscopy for the determination of the intramolecular distribution of 13C in glucose at natural abundance. Anal Chem 81: 8978-8985.
- Gilbert A, Silvestre V, Robins RJ, Remaud GS, Tcherkez G (2012) Biochemical and physiological determinants of intramolecular isotope patterns in sucrose from C3, C4 vand CAM plants accessed by isotopic 13C NMR spectrometry: a viewpoint. Nat Prod Rep 29: 476-486.
- Hayes JM (2001) Fractionation of Carbon and Hydrogen Isotopes in Biosynthetic Processes. Rev Mineral Geochem 43: 225-277.
- Meinschein WG, Rinaldi GG, Hayes JM, Schoeller DA (1974) Intramolecular isotopic order in biologically produced acetic acid. Biomed Mass Spectrom 1: 172-174.
- Rinaldi G, Meinschein WG, Hayes JM (1974) Intramolecular carbon isotopic distribution in biologically produced acetoin. Biomed Mass Spectrom 1: 415-417.
- Stanko RT, Tietze DL, Arch JE (1992) Body composition, energy utilization, and nitrogen metabolism with a severely restricted diet supplemented with dihydroxyacetone and pyruvate. Am J Clin Nutr 55: 771-776.
- Melzer E, Schmidt HL (1987) Carbon isotope effects on the pyruvate dehydrogenase reaction and their importance for relative carbon-13 depletion in lipids. J Biol Chem 262: 8159-8164.
- Melzer E, Schmidt HL (1988) Carbon isotope effects on the decarboxylation of carboxylic acids, comparison of the lactate oxidase reaction and the degradation of pyruvate by H2O2. Biochem J 252: 913-915.
- Bahl A, Bahl BS (2010) Advance organic chemistry. S Chand and Company Ltd.
- Chenier PJ (1992) Survey of Industrial Chemistry. 2nd revised edn. Wiley-VCH Publishers, New York, USA.
- Harold WA, Bryan GR (1996) Industrial Organic Chemicals. Wiley-Interscience, New York, USA.
- Japan External Trade Organization (2011) Guildbook for export to Japan (Food Articles).
- Zhang L, Kujawinski DM, Federherr E, Schmidt TC, Jochmann MA (2012) Caffeine in your drink: natural or synthetic? Anal Chem 84: 2805-2810.
- Suzuki Y, Akamatsu F, Nakashita R, Korenaga T (2010) A Novel Method to Discriminate between Plant- and Petroleum-derived Plastics by Stable Carbon Isotope Analysis. Chemistry Letters 39: 998-999.
- Calderone G, Guillou C (2008) Analysis of isotopic ratios for the detection of illegal watering of beverages. Food Chem 106: 1399-1405.
- Nimmanwudipong T, Gilbert A, Yamada K, Yoshida N (2015) Analytical method for simultaneous determination of bulk and intramolecular (13) C-isotope compositions of acetic acid. Rapid Commun Mass Spectrom 29: 2337-2340.
- Howard JW, Fraser WA (1932) Preparation of pyruvic acid. Org Synth Coll 1: 475-480.
- Serra F, Reniero F, Guillou CG, Moreno JM, Marinas JM, et al. (2005) 13C and 18O isotopic analysis to determine the origin of L-tartaric acid. Rapid Commun Mass Spectrom 19: 1227-1230.
- Zyakun AM, Oganesyants LA, Panasyuk AL, Kuz'mina EI, Shilkin AA, et al. (2015) Site-specific (13) C/(12) C isotope abundance ratios in dicarboxylic oxyacids as characteristics of their origin. Rapid Commun Mass Spectrom 29: 2026-2030.
- Rinaldi G, Meinschein WG, Hayes JM (1974) Carbon isotopic fractionations associated with acetic acid production by Acetobacter suboxydans. Biomed Mass Spectrom 1: 412-414.
- Li Y, Chen J, Lun SY (2001) Biotechnological production of pyruvic acid. Appl Microbiol Biotechnol 57: 451-459.
- Anton DL, Dicosmo R (1995) Witterholt Proccess for the preparation of pyruvic acid. WO patent, 95000656.
Relevant Topics
Recommended Journals
Article Tools
Article Usage
- Total views: 11422
- [From(publication date):
specialissue-2016 - Mar 31, 2025] - Breakdown by view type
- HTML page views : 10497
- PDF downloads : 925