Cognitive Impairment and Other Non-Motor Symptoms in Patients with Vascular Parkinsonism
Received: 03-Oct-2017 / Accepted Date: 06-Oct-2017 / Published Date: 13-Oct-2017 DOI: 10.4172/2161-0460.1000385
Abstract
Vascular parkinsonism (VP) is a rare cause of parkinsonism, however, it remains an important problem and need specific approach for diagnosis and therapy. Despite years of investigation in this field, there is no clarity in the definition and diagnosis criteria of VP. The modern concept of parkinsonian disorders declares the importance of non-motor symptoms in diagnosis and quality of life in patients. In this article, cognitive impairment and other non-motor symptoms of VP are considered.
Keywords: Vascular Parkinsonism; Vascular cognitive impairment; White matter lesions; Lacunes; Non-motor symptoms
Introduction
Vascular Parkinsonism (VP) is a rare variant of parkinsonism caused by ischemic or hemorrhagic lesions of basal ganglia (especially putamen or pallidum), midbrain, or their links with frontal lobes. According to different investigations, various forms of the cerebrovascular disease (CVD) may cause 1%-15% of Parkinsonism cases.
French neurologist was probably the first person who proposed the relation between vascular damage of substantia nigra and Some researchers found that Parkinsonism may be related to lesions of globus pallidus and putamen. In 1929, was published Critchley’s classic article “Arteriosclerotic parkinsonism” in the “Brain”, which contained the clinical description of VP [1]. Critchley divided “arteriosclerotic parkinsonism” into five clinical subtypes.
Later, Critchley [1] modified his conception and proposed the term “vascular pseudoparkinsonism” to highlight principal differences between neurodegenerative Parkinson’s disease (PD) and Parkinsonlike syndrome in CVD.
Other authors proposed to use the term “pseudovascular parkinsonism” in cases of imitation of vascular process (for example, in normal pressure hydrocephalus (NPH)) or “pseudovascular pseudoparkinsonism” in patients with other motor syndrome (for example, primary gait disorder, obsessional slowness, abulia, catatonia, paratonia, orparesis) that mimic some parkinsonian signs but also are caused by nonvascular lesions [2].
Boundaries of VP
CVD rarely manifests as the syndrome resembling classical parkinsonism of idiopathic PD or atypical parkinsonism (parkinsonismplus) associated with multiple system atrophy (MSA), progressive supranuclear palsy (PSP), and corticobasal degeneration (CBD). There are some difficulties in the definition of VP. At first, the coexistence of PD and CVD in elderly patients is not uncommon, while “true” VP occurs less often [3]. In most cases, CVD manifests as motor syndrome principally distinct from idiopathic PD or atypical parkinsonism. This motor syndrome is frequently named as “frontal gait disorder,” “frontal dysbasia,” “gait apraxia,” and sometimes “lower body parkinsonism” (reference to “Lewy-body parkinsonism” that is PD).
However, “lower body parkinsonism” is not a variant of the parkinsonian syndrome as not being in line with the main formula of parkinsonism: hypokinesia plus resting tremor and/or rigidity. Limb bradykinesia with decrement, being one of the diagnostic criteria of parkinsonism, is absent in patients with “lower body parkinsonism”. Furthermore, such patients have no obvious parkinsonian signs in the state of lying and sitting (so the correct term may be “walking parkinsonism,” not “lower body parkinsonism”).
“True” VP is a rare condition with a heterogeneous clinical picture which depends on the type of brain damage and location of the lesion. Vascular lesions non-selectively involve different functional systems, as a result, a complex constellation of different motor syndromes may be in one patient. Parkinsonian syndrome in patients with VP is prevailed by lower limb bradykinesia, gait disturbances, postural instability, absence or rare occurrence of resting tremor. It can also be associated with pyramidal signs, impairment of eye movements, pseudobulbar palsy, speech disorders, dysphagia, urinary incontinence and cognitive decline [4]. Typically, VP patients had an older age at onset of symptoms and shorter disease duration than PD patients [5].
The Pathophysiology of VP
VP is a syndrome and can be caused by different CVDs. Cerebral small vessel disease is the most frequent cause of the VP. Diffuse damage of penetrative long thin noncollateral arteries vascularized deep compartments of the brain (vascular “centrencephalon”) associated with long-term arterial hypertension is found in most cases of VP. Acute or subacute onset of VP can be related to infarction or other lesions affecting the basal ganglia. It is important that parkinsonism infrequently occurs in the recovery phase after stroke, thereby it replaces the symptoms of spastic paralysis. While strokes of the basal ganglia and deep white matter are very common in the elderly, most are not followed by parkinsonism. Indeed, silent infarcts in the basal ganglia were detected in 88 (40.2%) of 219 consecutive adults “requesting the medical evaluation for possible cerebrovascular diseases” [6]. In another study from 220 consecutive brain autopsies of patients with cerebral infarcts, only five had a clinical history of parkinsonian symptoms. In these five cases, there was no correlation between the location/size of the infarcts and the lateralization or severity of parkinsonism [7].
In the RUN DMC prospective study the risk of incident parkinsonism and dementia in 503 non-demented 50–85 years old participants with CVD was assessed [8,9]. After 5 years of follow-up 8.6% of participants developed dementia, while only 3.9% of patients had parkinsonism and 2.9% was diagnosed with VP. Thus, cognitive impairment occurs more often than parkinsonism in patients with CVD.
Radiological and post-mortem studies showed no correlation between leukoaraiosis and parkinsonism. Yamanouchi and Nagura [10] compared the brain pathology of 24 patients who had a premortem diagnosis of VP with 22 age-matched brains with CVD without parkinsonism. They found no significant difference in the extent of vascular lesions at the basal ganglia between two groups [10]. However, in another study, Bhatia and Marsden found that bilateral lesions of the lentiform nuclei, either of the globus pallidus or of the putamen, caused parkinsonism [11]. In our study, we compared MRI scans of 29 patients with VP and 26 patients with CVD without parkinsonism. Similarly, there was no significant difference in the extent of subcortical and periventricular white matter hyperintensities, lacunes or ventricular dilation. However, we found more prominent and frequent lesions in the putamen, globus pallidus and frontal lobes [4].
There are some implications in this and other studies. At first, there is no clear association between the burden of vascular pathology and parkinsonism. Secondly, as we conducted, ischemic or hemorrhagic lesions of area strategically located for parkinsonism plays a key role. VP can be divided into five main subtypes according to the main location of motor frontostriatal circuit disruption: (1) nigral (nigrostriatal), (2) striatopallidal, (3) frontostriatal, (4) thalamofrontal, and (5) mixed. Different variants are distinguished by the morphologic substrate (Figure 1 and Tables 1 and 2), pathophysiologic mechanism, proportionality of the main parkinsonian symptoms, relation of the axial and limbs signs, reaction to the dopaminergic drugs [4].
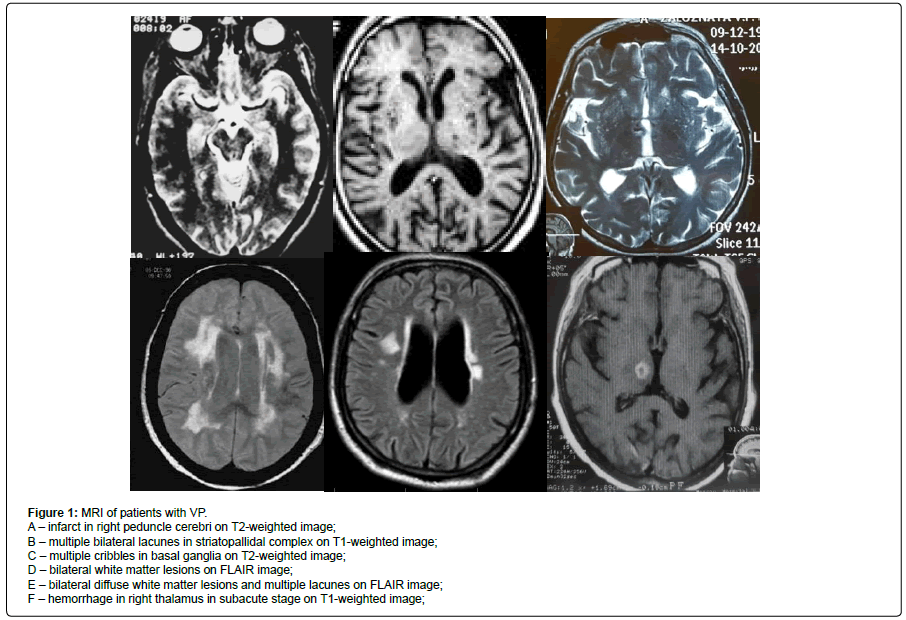
Figure 1: MRI of patients with VP.
A – infarct in right peduncle cerebri on T2-weighted image;
B – multiple bilateral lacunes in striatopallidal complex on T1-weighted image;
C – multiple cribbles in basal ganglia on T2-weighted image;
D – bilateral white matter lesions on FLAIR image;
E – bilateral diffuse white matter lesions and multiple lacunes on FLAIR image;
F – hemorrhage in right thalamus in subacute stage on T1-weighted image;.
| Types | Features |
|---|---|
| Type 1 | rigidity, hypomimia, marche apetits pas |
| Type 2 | pseudobulbar syndrome and variable pyramidal signs |
| Type 3 | dementia and incontinence |
| Type 4 | pallidopyramidal syndrome |
| Type 5 | pallidocerebellar syndrome |
Table 1: Clinical subtypes of “arteriosclerotic parkinsonism”.
| VP subtypes | Pathological substrate | Motor manifestation | Cognitive performance |
|---|---|---|---|
| Nigrostriatal | Ischemic and hemorrhagic lesions in midbrain affected nigrostriatal neurons; As a rule, bilateral |
Parkinsonism including postural, kinetic tremor, rest tremor (mesencephalic tremor); As a rule, good L-dopa response |
As a rule, relatively preserved cognition |
| Striatopallidal | Lacunar infarcts and microbleeds in basal ganglia; As a rule, bilateral |
Various parkinsonian features depending on location of vascular lesions, including rigidity, hypokinesia, gait disturbances, postural instability | As a rule, relatively preserved cognition |
| Frontostriatal | Diffuse white matter lesions or bilateral frontal infarcts | Symmetric parkinsonism, early freezing, postural instability; As a rule, lack of effect of L-dopa |
Frontal-subcortical type of cognitive impairment, high risk of dementia |
| Thalamofrontal | Ischemic and hemorrhagic lesions | Parkinsonism - extremely rare (in ventrolateral thalamic nucleus lesion) | Fluctuating dysexecutive cognitive deficit, sometimes associated with memory or verbal fluency impairment |
| Mixed | Diffuse multifocal lesions of frontostriatal circuits | Symmetric parkinsonism, gait disturbances, postural instability; lack of effect of L-dopa |
Frontal-subcortical type of cognitive decline up to dementia |
Table 2: Motor and cognitive features in VP subtypes.
Cognitive Impairment and Dementia in Patients with VP
Cognitive impairment is common manifestation of CVD which may be associated with different types of cerebrovascular lesions. Cognitive impairment in patients with CVD can be explained by selective damage of brain regions important for cognition and behaviour, especially executive functioning and attention. Regions emerging as especially vulnerable to advanced age and to essential hypertension include the prefrontal cortex, hippocampus and medial temporal cortex and inferior parietal lobule [12]. Moreover, white matter lesions disrupt cortical-subcortical circuits passing through the white matter and relate to the deficit in executive functions. Thus, the “critical” amount of white matter lesions can lead to cognitive decline and dementia.
In some studies the connection between cognitive impairment and different cerebrovascular lesions was revealed like multi-infarct lesions, lacunes and microhemorraghes, however others found that cognitive deficit is correlated with leukoaraiosis [13,14].
In most studies white matter hyperintensities were associated with a faster decline in global cognitive performance as well as in executive function and processing speed [13]. The significant correlation between subcortical and periventricular white matter hyperintensities (WMHs) and executive dysfunction in 170 patients with vascular mild cognitive impairment (MCI) [15]. The meta-analysis of 46 longitudinal studies showed that white matter hyperintensities increased the risk of cognitive decline and dementia by approximately two times [13].
According to some study reports, cognitive impairment is significantly more common in VP than in PD [4,5,16,17]. Cognitive decline was presented or developed early in the course of the disease in about 50% patients with VP. Levin [18] showed a higher percentage of dementia in patients with VP compared with PD patients.
Cognitive impairment is one of the most frequent, almost universal coexisting signs in patients with VP. PRIAMO Study Group (2010) found attention and memory impairment in 61 of 83 clinically diagnosed VP patients (73.5%). Vale et al. [19] reported that all 15 VP patients had cognitive impairment and 80% fulfilled diagnostic criteria for probable vascular dementia. In another study, probable vascular dementia was revealed in 70.5% of VP patients [20]. In the Bambuí study, 8 of 13 (61.5%) patients with VP presented with the concomitant diagnosis of vascular dementia [21]. Glass et al. [22] found concomitant dementia in 39% of the 28 patients with pathologically confirmed VP.
According to Marxreiter et al. [23], there is no correlation between the severity of VP and dementia. Moreover, although cognitive dysfunction correlates with the severity of leukoaraiosis, there is no such link according to parkinsonism that shows a lead role of lesion location in parkinsonism emergence.
From another point of view, parkinsonism may be additional criteria of the causative link between dementia/MCI and CVD. Thus, Boyle et al. [24] suggested that parkinsonian symptoms may be more prominent among individuals with nonamnestic MCI in contrast to amnestic MCI. Another study demonstrated that the severity of parkinsonian symptoms correlates with the severity of cognitive impairment, especially in non-memory domains [25]. Mauri et al. [26] investigated the association between mild parkinsonian symptoms and MCI and indicated for these cases a greater risk for an involvement of executive functions and the subsequent development of vascular dementia. Thus, the involvement of executive functions is consistent with the dysfunction of frontosubcortical systems and the emergence of parkinsonian signs [27]. Leukoencephalopathy and lacunar infarcts in the deep white matter of the frontal lobe, caudate, or putamen may cause dysfunction of frontostriatal circuits and thus induce both executive impairment and parkinsonian symptoms. Moreover, patients with cerebrovascular lesions may have more severe cognitive decline than patients with PD alone. Therefore, cerebrovascular lesions may have an important effect in determining severity and extent of cognitive dysfunction.
Dunet et al. [28] found that cognitive impairment in VP is associated with decreased functional relations between caudate nucleus and cingulate cortex. Thirty patients (8 with VP and 22 with PD) and 23 controls were investigated. Patients with VP had higher caudate nucleus and white matter hyperintensity lesion volumes. Caudate nucleus functional connectivity with the perigenual anterior cingulate cortex was increased in patients with VP compared with controls and patients with PD, and it was positively correlated with the caudate nucleus volume. Caudate nucleus functional connectivity with the posterior cingulate cortex was decreased in patients with VP compared with controls and negatively correlated with the number of errors on the Stroop test. These findings suggest that ischemia-related remodeling may contribute to cognitive decline in patients with VP.
Dementia in VP is usually frontal-subcortical, manifesting as a dysexecutive syndrome with impairment of attention, planning, judgment, goal-directed behavior, abstract thinking, verbal fluency, and early apathy. Santangelo et al. [16] found that although PD patients had an earlier onset and longer disease duration than VP patients, patients with vascular abnormalities had more severe executive dysfunction. This study and some other reports have found that executive deficits predominate in subjects with subcortical vascular pathology [29]. On the other side, Benítez-Rivero et al. [5] found that VP patients had a global pattern of cognitive impairment, including executive function, verbal memory, and language. Accordingly, patients with VP and cognitive impairment can be divided into three groups: 1) VP+dysexecutive MCI, 2) VP+global or amnestic MCI and 3) VP+vascular dementia. It is important that VP occurs less often than combination of VP and PD [30]. Thereby, CVD coexists with neurodegenerative process that explains different cognitive impairment profiles in such patients.
Other Non-Motor Symptoms in VP
Other non-motor symptoms are frequently found in VP patients and play a role in diagnosis of parkinsonism. The PRIAMO study found a high prevalence of psychiatric disturbances in patients with VP (77.1%) including apathy (42.2%) [31]. However, there are few data about affective symptoms in patients with VP. Much is written about “vascular depression hypothesis”, which suggests that CVD (especially silent cerebral small vessel disease) affects the frontolimbic system and induces late-onset depression [32-37]. There is not enough evidence supporting this hypothesis. However, LADIS showed that severity of white matter lesions predicts depressive symptoms, and supported its role in the pathogenesis of late-life depression [14].
According to Glass et al. [22], mood disorders were presented in 60.9% patients with pathologically confirmed VP. Some authors emphasize the specific links between apathy and severity of the motor disability. In the PRIAMO study, apathy was revealed in 42.2% of VP patients. Diagnostic significance of depression and other affective disorders in VP is low, but nevertheless, it is the important target for therapeutic management.
The patients with VP have an extremely low frequency of visual hallucinations compared with PD or DLB. So the presence of visual hallucinations makes the diagnosis of VP very unlikely [38]. On the other side found that delirium was more frequent in patients with degenerative dementia and coexisting CVD than in those without CVD. But there is no pathological confirmation of dementia etiology in this study. According to the PRIAMO study, psychiatric symptoms, attention, and memory impairment were more frequently observed in DLB patients than in VP.
It is known that autonomic dysfunction is common in PD affecting up to 80% of patients [39]. Autonomic dysfunction symptoms include gastrointestinal disorders, cardiovascular, genitourinary, thermoregulatory insufficiency and other aspects of the autonomic nervous system.
However, there are only a few studies exploring autonomic insufficiency in patients with VP. In one of them, Murata with colleagues assessed cardiovascular dysfunction in VP, PD and controls [40]. They investigated the relationship between blood pressure (BP), pulse rate (PR), the standing test, and the coefficient of variation in the R-R interval (CV[R-R]). The BP-PR relationship was lost in the patient groups that reflects an imbalance between sympathetic and parasympathetic nervous systems not only in PD but also in VP. Interestingly, the CV[R-R] in VP group was the lowest, exhibiting a sign of impaired parasympathetic function. Murata et al. found that the abnormal diurnal BP and PR variations are related to brainstem lesions due to ischemic changes or multiple infarctions in VP patients. Probably, lesions in the hypothalamus and in the locus coeruleus can cause preganglionic autonomic denervation and OH in patients with VP.
It is well established that cardiac [123I] metaiodobenzylguanidine (MIBG) uptake is significantly reduced in patients with PD at the early stages. This feature corresponds to the presence of myocardial postganglionic sympathetic dysfunction as part of the neurodegenerative process in PD. In patients with VP, cardiac MIBG uptake is substantially higher than in patients with PD and slightly lower than in controls [41]. This finding exhibits intact postganglionic sympathetic innervation in VP. In some studies, there was no association between cardiac sympathetic denervation and severity of OH. However, patients with Lewy body pathology with cerebral microbleeds showed less abnormal MIBG scans. Ghebremedhin et al. [42] revealed inverse relationship between vascular lesions and Lewy body pathology. Apparently vascular lesions may influence the course of the neurodegenerative process masking different features of the disease.
According to Fereshtehnejad and Lokk [43], symptomatic orthostatic hypotension (OH) is present in up to 30% patients with PD, 80% patients with MSA, 31% of DLB, and up to 26% patients with VP. OH increases the risk of postural instability and falls, which affects the quality of life in patients with different parkinsonian types. Postural symptoms due to OH were found in 18% of patients with VP. In several studies, it was shown that OH may be an intermediate link among WMHs and late-life depression [44].
Probably, connections between OH and VP can be more complicated. WMH due to cerebral blood flow autoregulation dysfunction may contribute to OH related symptoms in patients with CVD including light-headedness, fatigue, blurred vision, presyncope and even syncope. Moreover, WMH volume is believed to be associated with the severity of postural symptoms. Transient cerebral hypoperfusion may mediate the association between OH and cognitive impairment. The causal link between OH and cognitive impairment remains a subject of debate. Frequently the presence of OH and cognitive impairment in patients with VP may reflect neurodegenerative process. Although subject to the absence of neurodegeneration OH negatively influences cognition including mediating impact of aggravating WMH creating a “circulus vitiosus”. Longitudinal neuroimaging studies may help clarify the nature of this relationship. Furthermore, early diagnosis and treatment of OH may improve the prognosis in these patients.
Gastrointestinal (GI) dysfunction was found in 65%-80% of patients with VP in different studies. According to Park et al. [45], the frequency of GI dysfunction in 62 patients with VP was similar to PD and other parkinsonian disorders. Constipation was the most common symptom, followed by appetite loss, weight loss, dysphagia, and sialorrhea.
Urinary symptoms were found in 65%–90% of patients with VP. In the PRIAMO study, GI symptoms, pain, urinary problems, and postural instability due to OH were most frequent in MSA than in VP, whereas the prevalence of skin and respiratory disorders was rather low in all forms of parkinsonism, ranging between 10% and 30% (for example, respiratory symptoms were found in 25% of patients with VP).
According to the PRIAMO study, sleep disturbances were found in 70% of patients with VP and other parkinsonism subtypes (except CBD-36%) [31]. Similar to PD, patients with VP identified nocturia and the problem of getting up at night to urinate as being the most frequent and troublesome nocturnal symptom, but nocturnal akinesia is not a typical symptom in VP. In total, PD patients in comparison with VP patients reported significantly more troublesome problems with sleep quality [46]. VP patients have higher score sleep domain compared to PD patients (18.7 ± 8.5 vs. 15.8 ± 8.3) [47].
It is well known that rapid eye movement sleep behavior disorder (RBD) is strongly correlated with synucleinopathies. Iranzo et al. [48] found RBD in 8.9% of patients with vascular cognitive impairment and in 4.2% of patients with stroke. Patients with CVD also showed a higher prevalence of restless legs syndrome (RLS)-16.1%, and obstructive sleep apnea syndrome (35.7%). Polysomnography studies revealed the reduced proportion of slow-wave sleep and rapid eye movement sleep, prolonged sleep latency, and decreased sleep efficiency index. Subjective sleep disturbances included difficulties in falling asleep, waking early, sleep fragmentation, and RLS. Two basic mechanisms might be hypothesized for the generation of sleep disturbances in these patients [49]: (1) vascular damage influences directly on neural structures involved in sleep regulation, affecting the normal sleep-wake system and then determining sleep structure disruption [50]; (2) CVD determines a central imbalance of neurotransmitters, such as serotonin, melatonin, and acetylcholine, causing sleep structure abnormalities [51]. In addition, it was shown that patients with RBD had abnormal MIBG-scans, that apparently may differentiate PD from VP.
It is established that most of the patients with PD have impaired olfactory function. In VP, hyposmia was found in 39% of patients [31]. Navarro-Otano et al. [52] found that patients with suspected VP had lower UPSIT score than in controls but higher than in PD patients. Patients with low cardiac [53] I-MIBG score suggestive of PD were more likely to have a higher UPSIT score. In addition, Katzenschlager et al. [54] also found that UPSIT scores in VP were significantly better than in PD and did not differ from the healthy controls. Only 4 of the 14 (28.6%) patients with VP and 7 of the 27 (25.9%) controls had scores below published optimal cutoff values for the respective age and sex groups. Katzenschlager et al. [54] suggested a cutoff value ≤ 23 showing the best balance between sensitivity and specificity in the 65–75 group, with 100% sensitivity and a specificity of 85.7%. In the 76–88 group, UPSIT score of ≤ 22 yielded a sensitivity of 85.7% and a specificity of 80%.
Olfactory dysfunction corresponds to findings of Lewy bodies in the anterior olfactory nucleus, showing the involvement of the olfactory system in the neurodegenerative process. Studies showed that hyposmia occurs in patients with VP, but its prevalence is much less than that in PD.
Diagnostic Criteria of VP
The appearance of CT and MRI made the diagnosis of CVD simpler but formed “diagnostic trap” with the desire to explain any neurological deficit by vascular lesions found. Furthermore, cerebrovascular lesions are a common incidental finding in pathologically confirmed PD. So, diagnosis of VP must be only the “diagnosis of exclusion”.
Jellinger [55] found that different variants of vascular damage of the brain (multi-infarct state, leukoencephalopathy, multiple lacunar infarcts in basal ganglia and brainstem) appear to be the cause of parkinsonism in 6% of cases, but epidemiologic data showed the higher prevalence of VP (8%-12%). Combination of different forms of CVD and Lewy-body pathology occurs 4 times more often than pure VP.
Proposed criteria for VP diagnosis include: (1) parkinsonian syndrome defined as hypokinesia plus rigidity and/or resting tremor; (2) CVD objectively supported by supplemental diagnostic methods such as MRI, MR-angiography, CT-perfusion, CT-angiography, and ultrasound; and (3) causal links between 1 and 2 confirmed by clinical features or clinical-imaging correlations.
Clinical features of VP include an absence of resting tremor, predominance of parkinsonian symptoms in lower extremities and axial region, earlier emergence of postural instability and gait disturbances, a bilateral onset of the disease, and low efficacy of dopaminergic drugs. There are no any pathognomonic signs, which confirmed the diagnosis of VP. The pronounced dissociation between the involvement of the legs and axial muscles and the intact movements of the hands has no adequate explanation. It can be assumed that basal ganglia lesions cause dysfunction of subcortical-brainstem pathways which control axial muscles, which provide balance and body-extremity synergism via the medial brainstem-spinal system.
Frequently, VP has a slowly progressive course, but in 20%–25% of cases, the course of the disease can be acute or subacute (usually 1–6 months after stroke).
Diagnosis of VP is less likely in the following cases: (1) absence of both vascular lesions and lesions in strategic areas on MRI; (2) high persistent L-dopa effect throughout 5 years; (3) worsening of parkinsonism in levodopa withdrawal; (4) downgaze paralysis in the absence of upgaze paralysis; (5) progressive autonomic dysfunction; (6) early emergence of visual hallucinations; (7) atrophy of midbrain or putamen; or other MRI-signs of atypical parkinsonism; (8) emergence of motor fluctuations and dyskinesia.
Treatment
There are two main directions for treatment of VP: (1) course disease modification through the reduction of vascular risk factors and (2) symptomatic treatment. Common risk factors for VP are same as those for CVD, and their prevention and treatment are of utmost importance. Hypertension is prevailing among treatable vascular risk factors, like as diabetes, metabolic syndrome, hyperlipidemia, hyperhomocysteinemia, atrial fibrillation and smoking. Another way to influence progression of VP is the antiplatelet therapy which prevents lacunar infarction. However, it is not known if this approach ameliorates motor and cognitive symptoms in these patients.
The only accepted symptomatic treatment for VP is levodopa. The levodopa response in VP varies from 20-40% [56]. ? poor or non-sustained response to levodopa is another differentiating feature between VP and PD. However, an attempt of levodopa use should be done in all patients. A dose of up to 1000 mg/day should be given for at least 3 months before labeling the patient as a non-responder or poor responder. In good responders, the duration of sustained response is typically less than in PD, with the drug becoming non-beneficial after 1or 2 years. The above response pattern occurs in VP because of the location of lesions in VP. Within VP, patients with lesions (infarcts) in or near nigrostriatal pathway have a higher chance of responding to levodopa than those who have a greater white matter disease [17,57]. Patients with lesions in or near the basal ganglia may show a poor uptake of ligand in [123I] FP-CIT DAT-SPECT, and thus DAT-SPECT may guide the treatment in VP [58].
In a study with quantitative assessment of motor symptoms in VP and PD, it was shown that the improvement in motor function after levodopa was 19% in patients with VP, while patients with PD had more than 50% improvement [59].
Chronic levodopa response is positive in 48% and negative in 52% of patients with VP. A negative response to levodopa was associated with greater frequency of symmetric onset of motor symptoms, worst disease severity, the absence of dyskinesia and the greater number of vascular risk factors. Frontal lobe showed largest vascular load. Striatal DAT was normal in 30% and abnormal in 70% of patients. Patients with normal DAT binding showed higher vascular load at MRI. Significant predictive factors of worst disease severity and negative response to levodopa were hypertension, vascular lesions in basal ganglia/ periventricular regions, and normal DAT uptake [60].
The therapy of cognitive impairment in VP as a whole is ineffective. Acetylcholinesterase inhibitors and the NMDA receptor antagonist memantine have been shown to slightly ameliorate executive functions in patients with VP. In general, the effect of cognition-enhancing drugs is not so strong to change the level of daily activities and cognitive status impression.
Conclusion
VP and vascular cognitive impairment are two main clinical features of CVD. The interrelation of motor and cognitive symptoms is complex and related on location of vascular lesions. As a result, a complex constellation of different motor and non-motor syndromes may be seen in one patient.
“True” VP is a rare condition and can be diagnosed only in the presence of parkinsonism. Cognitive impairment with a predominance of executive dysfunction more often accompanies motor disorders in patients with VP, than with PD. Other non-motor symptoms are less frequent in VP than in PD. Thereby, the presence or absence of specific non-motor symptoms can help distinguish bridges and boundaries in cerebrovascular and neurodegenerative diseases.
The treatment of patients with VP should include course disease modification through the reduction of vascular risk factors. Levodopa and cognition-enhancing drugs have variable effect but should be tried in every patient with motor and cognitive symptoms.
References
- Vizcarra JA, Lang AE, Sethi KD, Espay AJ (2015) Vascular parkinsonism: Deconstructing a syndrome. Mov Disord 30: 886-894.
- Levin OS (2003) Movement disorders in cerebrovascular diseases. In: Extrapyramidal disorders. Diagnosis and treatment by Shtok VN (eds). Moscow: MedPress-In- form, 503-519.
- BenÃtez-Rivero S, Lama MJ, Huertas-Fernández I, de Toledo PÃ, Cáceres-Redondo MT, et al. (2014) Clinical features and neuropsychological profile in vascular parkinsonism. J Neurol Sci 345: 193-197.
- Uehara T, Tabuchi M, Mori E (1999) Risk factors for silent cerebral infarcts in subcortical white matter and basal ganglia. Stroke 30: 378-382.
- De Reuck J, Sieben G, De Coster W, Vander Eecken H (1980) Parkinsonism in patients with cerebral infarcts. Clin Neurol Neurosurg 82: 177-185.
- van der Holst HM, van Uden IW, Tuladhar AM, de Laat KF, van Norden AG, et al. (2015) Cerebral small vessel disease and incident parkinsonism The RUN DMC study. Neurology 85: 1569-1577.
- van Uden IW, van der Holst HM, Tuladhar AM, van Norden AG, de Laat KF, et al. (2016) White matter and hippocampal volume predict the risk of dementia in patients with cerebral small vessel disease: the RUN DMC study. J Alzheimers Dis 49: 863-873.
- Yamanouchi H, Nagura H (1997) Neurological signs and frontal white matter lesions in vascular parkinsonism. Stroke 28: 965-969.
- Bhatia KP, Marsden CD (1994) The behavioural and motor consequences of focal lesions of the basal ganglia in man. Brain 117: 859-876.
- Gasecki D, Kwarciany M, Nyka W, Narkiewicz K (2013) Hypertension, brain damage and cognitive decline. Curr Hypertens Rep15: 547-558.
- Debette S, Markus HS (2010) The clinical importance of white matter hyperintensities on brain magnetic resonance imaging: Systematic review and meta-analysis. Bmj 341: c3666.
- The LADIS Study Group, Poggesi A, Pantoni L, Inzitari D, Fazekas F, Ferro J, et al. (2011) 2001-2011: A decade of the LADIS (Leukoaraiosis and DISability) study: What have we learned about white matter changes and small-vessel disease? Cerebrovasc Dis 32: 577-588.
- Bombois S, Debette S, Delbeuck X, Bruandet A, Lepoittevin S, et al. (2007) Prevalence of subcortical vascular lesions and association with executive function in mild cognitive impairment subtypes. Stroke 38: 2595-2597.
- Santangelo G, Vitale C, Trojano L, De Gaspari D, Bilo L, et al. (2010) Differential neuropsychological profiles in parkinsonian patients with or without vascular lesions. Mov Disord 25: 50-56.
- Zijlmans JCM, Katzenschlager R, Daniel SE, Lees AJL (2004) The L-dopa response in vascular parkinsonism. J Neurol Neurosurg Psychiatry 75: 545-547.
- Vale TC, Caramelli P, Cardoso F (2015) Clinicoradiological comparison between vascular parkinsonism and Parkinson's disease. J Neurol Neurosurg Psychiatry 86: 547-553.
- Vale TC, Caramelli P, Cardoso F (2013) Vascular parkinsonism: a case series of 17 patients. Arq Neuropsiquiatr 71: 757-762.
- Barbosa MT, Caramelli P, Maia DP, Cunningham MC, Guerra HL, et al. (2006) Parkinsonism and Parkinson's disease in the elderly: A community-based survey in Brazil (the Bambuà study). Mov Disord 21: 800-808.
- Glass PG, Lees AJ, Bacellar A, Zijlmans J, Katzenschlager R, et al. (2012) The clinical features of pathologically confirmed vascular parkinsonism. J Neurol Neurosurg Psychiatry 83: 1027-1029.
- MarxreiterF,WinklerJ(2016)VaskuläresParkinsonSyndrom(ParkinsonismusbeivaskulärerEnzephalopathie). Fortschr Neurol Psychiatr 84: 8-13.
- Boyle PA, Wilson RS, Aggarwal NT, Arvanitakis Z, Kelly J, et al. (2005) Parkinsonian signs in subjects with mild cognitive impairment. Neurology 65: 1901-1906.
- Rozzini L, Chilovi BV, Bertoletti E (2007) Mild parkinsonian signs and psycho-behavioral symptoms in subjects with mild cognitive impairment. International Psychogeriatrics 17: 1-10.
- Mauri M, Corbetta S, Pianezzola C, Ambrosoni E, Riboldazzi G, et al. (2008) Progression to vascular dementia of patients with mild cognitive impairment: Relevance of mild parkinsonian signs. Neuropsychiatr Dis Treat 4: 1267-1271.
- Wilson RS, Schneider JA, Beckett LA, Evans DA, Bennett DA (2002) Progression of gait disorder and rigidity and risk of death in older persons. Neurology 58: 1815-1819.
- Dunet V, Deverdun J, Charroud C, Le Bars E, Molino F, et al. (2016) Cognitive impairment and basal ganglia functional connectivity in vascular parkinsonism. AJNR Am J Neuroradiol 37: 2310-2316.
- Gorelick PB, Scuteri A, Black SE, DeCarli C, Greenberg SM, et al. (2011) Vascular contributions to cognitive impairment and dementia: A statement for healthcare professionals from the american heart association/american stroke association. Stroke 42: 2672–2713.
- Jellinger K (2007) Morphological substrates of parkinsonism with and without dementia: A retrospective clinico-pathological study. J Neural Transm Suppl 72: 91-104.
- Colosimo C, Morgante L, Antonini A, Barone P, Avarello TP, et al. (2010) Non-motor symptoms in atypical and secondary parkinsonism: The PRIAMO study. Journal of Neurology 257: 5-14.
- Alexopoulos GS, Meyers BS, Young RC, Campbell S, Silbersweig D, et al. (1997) ‘Vascular depression’ hypothesis. Arch Gen Psychiatry 54: 915-922.
- Alexopoulos GS, Meyers BS, Young RC, Kakuma T, Silbersweig D, et al. (1997) Clinically defined vascular depression. Am J Psychiatry 154: 562-565.
- Baldwin RC, O'Brien J (2002) Vascular basis of late-onset depressive disorder. Br J Psychiatry 180: 157-160.
- Taylor WD, Aizenstein HJ, Alexopoulos GS (2013) The vascular depression hypothesis: Mechanisms linking vascular disease with depression. Mol Psychiatry 18: 963-974.
- White CL, McClure LA, Wallace PM, Braimah J, Liskay A, et al., (2011) The correlates and course of depression in patients with lacunar stroke: Results from the secondary prevention of small subcortical strokes (SPS3) study. Cerebrovasc Dis 32: 354-360.
- Wu RH, Li Q, Tan Y, Liu XY, Huang J (2014) Depression in silent lacunar infarction: A cross-sectional study of its association with location of silent lacunar infarction and vascular risk factors. Neurol Sci 35: 1553-1559.
- Williams DR, Warren JD. Lees AJ (2008) Using the presence of visual hallucinations to differentiate Parkinson's disease from atypical parkinsonism. J Neurol Neurosurg Psychiatry 79: 652-655.
- Zesiewicz TA, Baker MJ, Wahba M, Hauser RA (2003) Autonomic nervous system dysfunction in Parkinson’s disease. Curr Treat Options Neurol 5: 149-160.
- Murata Y, Harada T, Ishizaki F, Izumi Y, Nakamura S (1997) Autonomic dysfunction in Parkinson's disease and vascular parkinsonism. Acta Neurol Scand 96: 359-365.
- Kim JS, Lee PH, Lee KS, Park JW, Kim YI, et al. (2006) Cardiac [123I] metaiodobenzylguanidine scintigraphy for vascular Parkinsonism. Mov Disord 21: 1990-1994.
- Ghebremedhin E, Rosenberger A, Rub U, Vuksic M, Berhe T, et al. (2010) Inverse relationship between cerebrovascular lesions and severity of lewy body pathology in patients with lewy body diseases. J Neuropathol Exp Neurol 69: 442-448.
- Fereshtehnejad SM, Lökk J (2014) Orthostatic hypotension in patients with Parkinson's disease and atypical parkinsonism. Parkinsons Dis 2014: 475854.
- Colloby SJ, Vasudev A, O'Brien JT, Firbank MJ, Parry SW, et al. (2011) Relationship of orthostatic blood pressure to white matter hyperintensities and sub-cortical volumes in late-life depression. Br J Psychiatry 199: 404-410.
- Park H, Lee JY, Shin CM, Kim JM, Kim TJ, et al. (2015) Characterization of gastrointestinal disorders in patients with parkinsonian syndromes. Parkinsonism Relat Disord 21: 455-460.
- Bhidayasiri R, Jitkritsadakul O, Petchrutchatachart S, Kaewwilai L, Panyakaew P, et al. (2014) Nocturnal manifestations of atypical and vascular parkinsonism: How do they differ from Parkinson's disease? J Neural Transm (Vienna) 121: 69-77.
- Pan M, Gao H, Long L, Xu Y, Liu M, et al. (2013) Serum uric acid in patients with Parkinson's disease and vascular parkinsonism: a cross-sectional study. Neuroimmunomodulation 20: 19-28.
- Iranzo A, Fernández-Arcos A, Tolosa E, Serradell M, Molinuevo JL, et al. (2014) Neurodegenerative disorder risk in idiopathic REM sleep behavior disorder: Study in 174 patients. PLoS One 9: e89741.
- Ahlberg J, Norlen L, Blomstrand C, Wikkelsö C (1988) Outcome of shunt operation on urinary incontinence in normal pressure hydrocephalus predicted by lumbar puncture. J Neurol Neurosurg Psychiatry 51: 105-108.
- Ahn HJ, Yoo WK, Park J, Ma HI, Kim YJ (2015) Cognitive dysfunction in drug induced parkinsonism caused by prokinetics and antiemetics. J Korean Med Sci 30: 1328-1333.
- Bassetti CL, Hermann DM (2011) Sleep and stroke. Handb Clin Neurol 99: 1051-1072.
- Navarro-Otano J, Gaig C, Muxi A, Lomeña F, Compta Y, et al. (2014) 123 I-MIBG cardiac uptake, smell identification and 123 I-FP-CIT SPECT in the differential diagnosis between vascular parkinsonism and parkinson's disease. Parkinsonism Relat Disord 20: 192-197.
- Tullberg M, Hellstrom P, Piechnik SK, Starmark JE, Wikkelso C (2004) Impaired wakefulness is associated with reduced anterior cingulate CBF in patients with normal pressure hydrocephalus. Acta Neurol Scand 110: 322-330.
- Katzenschlager R, Zijlmans J, Evans A, Watt H, Lees AJ (2004) Olfactory function distinguishes vascular parkinsonism from Parkinson's disease. J Neurol Neurosurg Psychiatry 75: 1749-1752.
- Jellinger K (2005) Vascular parkinsonism. Comments on Sibon et al. J Neurol (2004) 251:513-524. J Neurol 252: 1549.
- Constantinescu R, Richard I, Kurlan R (2007) Levodopa responsiveness in disorders with parkinsonism: A review of the literature. Mov Disord 22: 2141-2148.
- Gupta D, Kuruvilla A (2011) Vascular parkinsonism: What makes it different? Postgrad Med J 87: 829-836.
- Zijlmans J, Evans A, Fontes F, Katzenschlager R, Gacinovic S, et al. (2007) [123I] FP-CIT spect study in vascular parkinsonism and Parkinson's disease. Mov Disord 22: 1278-1285.
- Gago MF, Fernandes V, Ferreira J, Silva H, Rodrigues ML, et al. (2015) The effect of levodopa on postural stability evaluated by wearable inertial measurement units for idiopathic and vascular Parkinson's disease. Gait Posture 41: 459-464.
- Antonini A, Vitale C, Barone P, Cilia R, Righini A, et al. (2012) The relationship between cerebral vascular disease and parkinsonism: The VADO study. Parkinsonism Relat Disord 18: 775-780.
Citation: Levin OS, Chimagomedova A, Iakovleva OV, Skripkina NA, Lyashenko EA (2017) Cognitive Impairment and Other Non-Motor Symptoms in Patients with Vascular Parkinsonism. J Alzheimers Dis Parkinsonism 7: 385. DOI: 10.4172/2161-0460.1000385
Copyright: © 2017 Levin OS, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Share This Article
Recommended Journals
Open Access Journals
Article Tools
Article Usage
- Total views: 7013
- [From(publication date): 0-2017 - Apr 19, 2025]
- Breakdown by view type
- HTML page views: 6106
- PDF downloads: 907