Autophagy as a Possible Target for Cancer Therapy
Received: 09-Apr-2018 / Accepted Date: 27-Apr-2018 / Published Date: 03-May-2018 DOI: 10.4172/2472-016X.1000124
Abstract
Autophagy is an evolutionarily conserved catabolic process that targets cellular organelles and cytoplasmic constituents to the lysosomes for degradation. According to the type of cargo delivery, there are three main types of autophagy systems in mammals: macroautophagy, microautophagy, and chaperone-mediated autophagy. Macroautophagy, often simply (and hereafter) referred to as autophagy, is the best studied autophagic process and focus of this review. Autophagy has been recognized to be a pro-survival mechanism at times of cellular stress including starvation. Besides, increasing evidence indicated the importance of autophagy in in the pathogenesis of several diseases including cancer. But, its role in cancer is more complex and still controversial; it appears to be tumor suppressive during tumorigenesis, but contributes to tumor cell survival during cancer progression. Besides, autophagic capacity was shown to significantly affect responses of cancer cells to anticancer agents and radiation. Even though there is still a gap about how autophagy is regulated in cancer, it appears to provide a promising target for cancer treatment. This review aimed at examining the multiple roles of autophagy as a novel target for cancer therapy..
Introduction
Autophagic responses can be relatively non-selective or, on the contrary, highly specific. In this regard, non-selective autophagy executed at times of nutrient deprivation and ones activated several intracellular entities could be degraded to meet metabolic demand of cells by generating metabolic precursor for cell survival [1]. Whereas, selective autophagy is triggered in response to specific cellular homeostatic needs [2]. For instance, ribophagy elicited in order to selectively degrade defective ribosomes, while mitophagy make sure the selective degradation of defective mitochondria, and there are also other selective autophagic process targeted against other cellular components and cellular aggregates and intracellular bacteria and virus [3].
The molecular basis of autophagy has been well studied, mainly in yeasts, through examination of autophagy-defective mutants to identify the responsible genes (designated as AutophaGy; ATG), and according to current reports about 35 ATG genes have been discovered in yeast. More than half of these genes have obvious mammalian counterparts, and many of the core aspects of the process are conserved [1]. During autophagic process ATG proteins are sequentially activated to regulate different stages of autophagy. These proteins can be grouped into various complexes based on their functions: the uncoordinated-51- like kinase (ULK) complex, the ATG9-ATG2-WIPI1/ATG18 complex, class III phosphatidylinositol 3-kinase (PI3KIII) complex, and 2 ubiquitin-like conjugation systems, which include the ATG12–ATG5 and microtubule-associated protein I light chain 3 (LC3)/γ amino butyric acid A receptor-associated protein-like 1 (GABARAP) proteins. The fundamental sequential steps of process (Figure 1) includes induction, nucleation of membrane, elongation and maturation, fusion and, degradation of cargo contents by lysosome, recycling and autophagic lysosome reformation [4-8]. Furthermore, several signalling pathways regulate autophagy in mammalian cells (Figure 2), including phosphoinositide 3-kinase/v-akt murine thymoma viral oncogene homolog/mTOR-C1 (PI3K/AKT/mTOR-C1), adenosine monophosphate activated protein kinase (AMPK), mitogen-activated protein kinase (MAPK) pathways [9-17]. To this end this review aimed at scrutinizing the multiple roles of autophagy as a novel target for cancer therapy.
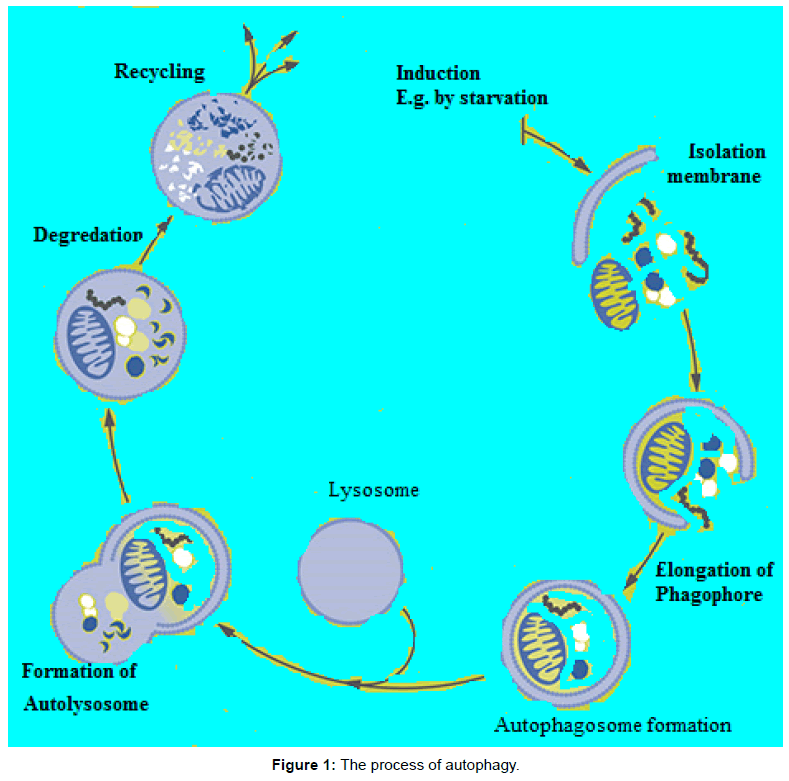
Figure 1: The process of autophagy.
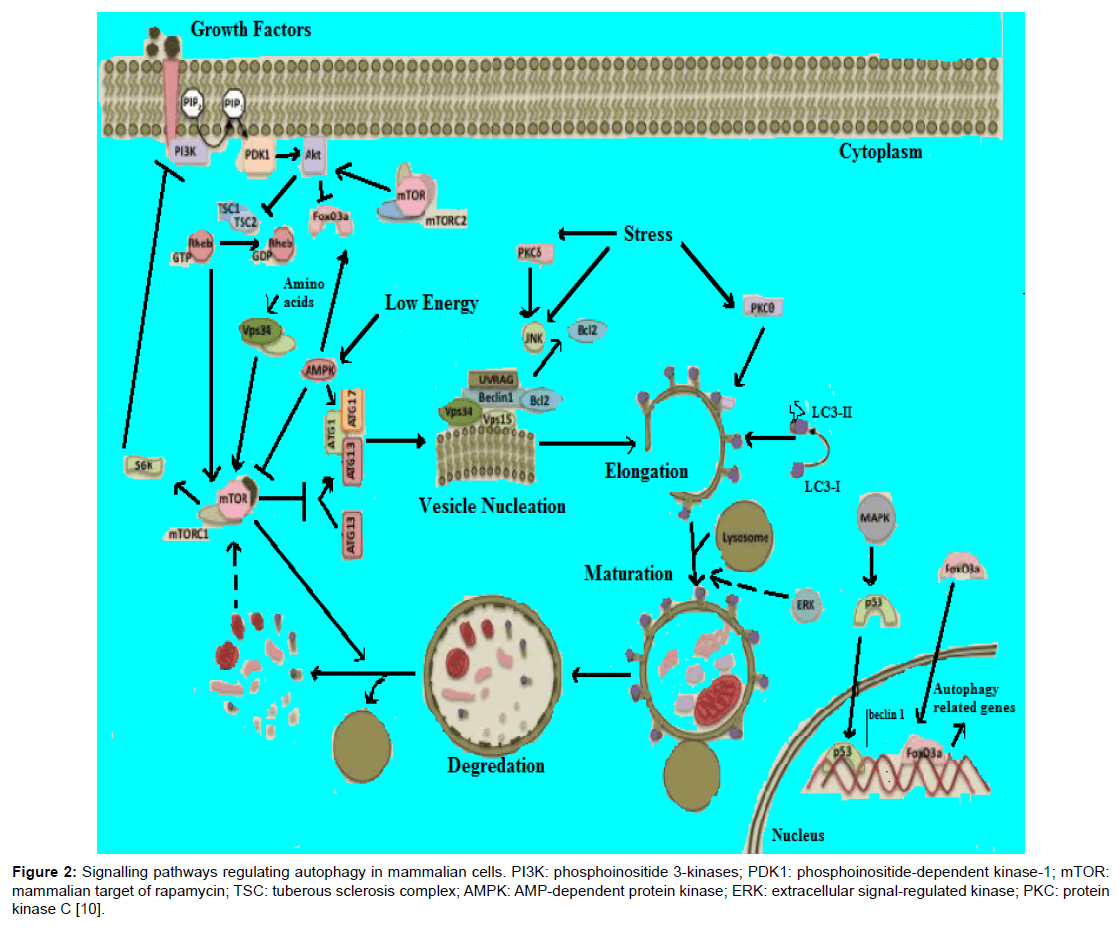
Figure 2: Signalling pathways regulating autophagy in mammalian cells. PI3K: phosphoinositide 3-kinases; PDK1: phosphoinositide-dependent kinase-1; mTOR: mammalian target of rapamycin; TSC: tuberous sclerosis complex; AMPK: AMP-dependent protein kinase; ERK: extracellular signal-regulated kinase; PKC: protein kinase C [10].
Role in Tumor Development and Progression
There is now increasing evidence that autophagy has complex and paradoxical roles in tumorigenesis, tumor progression and cancer therapeutics. Currently, there is a consensus that tumor suppressive functions of autophagy act during tumor initiation, and as a survival strategy by established tumors to cope with diverse stresses of the microenvironment that are encountered during tumor progression and metastasis [4,18-20].
Tumor Suppression
Early studies indicated that malignant or transformed cells often display lower basal autophagic activity than their normal counterparts [21,22]. This finding in differential expression suggests a link between tumorigenesis and decreased levels of autophagy. Furthermore, the identification of the genes required for the process offers opportunity to use genetic approaches to explore the role of autophagy in cancer development. From early studies, it was observed that Becn1 is monallelically deleted in around 50% of breast, ovarian and prostate cancers [23,24]. Moreover, one genetic study revealed that mice homozygously deleted for Beclin1 die during embryogenesis [25]. However, subsequent studies using mice with heterozygous deletion of Beclin1 showed that increased frequency of spontaneous cancers including lung adenocarcinomas, hepatocellular carcinomas (HCC), and lymphomas [25,26]. Thus, these studies provided the first direct genetic evidence that Beclin1 functions as a haplo insufficient tumor suppressor. In addition, other study findings also suggested the role of autophagy regulator, Beclin 1, in these in vitro and in vivo studies ectopic over expression of Beclin1 in Beclin 1 deficient mammary carcinoma cells result in reduction in tumor cell proliferation and also reduced tumorigenic potential in vivo, further suggesting a role for this autophagy regulator in tumor suppression [27,28]. Like Beclin1, UVRAG is mono-allelically deleted in human colon carcinoma [29]. Whereas, in human gastric carcinomas a frame-shift mutations in poly (A) tail of UVRAG gene has been documented in a study done by Kim et al. [30] that resulted in reduction in autophagy activity in these tumor cells. In addition to the above, tumor-associated deletions or mutations have been found in a number of other autophagy regulators as shown in Table 1.
| Gene/protein | Cancer-related changes |
|---|---|
| ULK1 | ↑ed expression in esophageal squamous cell & HCC; ↓ed expression in breast cancers |
| ATG2B | Frameshift mutations in gastric & colorectal cancers |
| ATG3 | ↓ed expression in myelodysplastic syndrome patients with leukemic evolution |
| ATG4B | ↑ed expression in CD34(+) chronic myeloid leukemia cells |
| ATG5 | Low-frequency frameshift mutations in gastric & colorectal cancers; Genetic variations may correlate with thyroid carcinoma susceptibility; ↓ed expression in natural killer cells & in gastric, colorectal, & HCC; ↑ed expression in CD34(+) chronic myeloid leukemia cells |
| LC3 | ↑ed expression in triple-negative breast cancer, colorectal & pancreatic cancers, & non-Hodgkin lymphomas; ↓ed expression in lung cancers, melanomas, & glioblastomas |
| ATG9B | Frameshift mutations in gastric & colorectal cancers |
| ATG10 | ↑ed expression in colorectal cancers is associated with metastasis; Genetic variations may correlate with breast cancer susceptibility |
| ATG12 | Frameshift mutations in gastric & colorectal cancers |
| ATG16L1 | Genetic variations may correlate with colorectal & thyroid cancers susceptibility; ↑ed expression in oral squamous cell carcinomas |
| BIF1 | ↓ed expression in pancreatic ductal adenocarcinomas, colorectal, gastric, urinary bladder, gallbladder, & prostate cancers |
| BECN1 | ↓ed expression in breast cancers, non-small cell lung cancers, renal clear cell carcinomas, brain tumors, cervical squamous cell, HCC, ovarian cancers, osteosarcomas, melanomas, & glioblastomas; Mutations in ovarian, human breast, prostate cancers; ↓ed expression due to gene methylation in breast cancers; ↑ed expression in CD34(+) chronic myeloid leukemia cells, colon cancers (stage IIIB), non-Hodgkin lymphomas, cholangiocarcinomas |
| UVRAG | Mutations in colorectal & gastric cancers |
| PIK3C/hVPS34 | Differentially expressed b/n the TEL/AML1-positive & negative acute lymphocytic leukemia groups; SNP associated with esophageal squamous cell carcinomas |
| GABARAP | ↑ed expression in colorectal carcinomas & benign & malignant thyroid tumors; ↓ed expression in neuroblastomas & breast cancers |
| GABARAPL1 | ↓ed expression in lymph node-positive high-grade breast cancers, acute myelocytic leukemias, & HCC |
| GABARAPL2 | ↓ed expression in acute myelocytic leukemias |
Table 1: Cancer-related changes in mammalian core autophagy genes and proteins.
Not only do mutations of the autophagy gene promote tumorigenesis, but autophagy is also positively regulated by the tumor suppressor genes and negatively regulated by the oncogenic pathways. Oncogenes like Akt and Ras inhibit autophagy primarily by activating the mTOR signaling pathway. Conversely, tumor suppressor PTEN which inhibit PI3K/Akt/mTOR-C1 pathway can activate autophagy [11,31]. Therefore, mutations in PTEN result in constitutive activation of the pathway, suppression of autophagy, and may contribute to tumor formation [6]. Other tumor suppressors such as TSC1, TSC2, p53, and liver kinase B1 (LKB1) stimulate autophagy through their inhibitory effects on mTOR-C1 [10]. To this end, number mechanisms could clarify the tumor suppressive roles of autophagy, including prevention of oxidative stress and genomic instability, inhibition of necrosis and inflammation, promotion of cancer cell death, modulation of antitumor immune response, maintenance of normal stem cells and degradation of oncogenic proteins [4].
Prevention of Oxidative Stress and Genomic Instability
It has been known that, during carcinogenesis there is a reduction or inactivation of apoptosis, which regarded as the primary mechanism involved in clearing defective cells. In this case cells greatly relay on autophagy to maintain cellular fitness. A study done by Mathew et al. using baby mouse kidney epithelial cells immortalized from mutant mice (iBMK cells) reported that defects in autophagy, associated with either allelic loss of beclin1 or lack of ATG5, result in diminished survival during metabolic stress and a heightened DNA damage response. Moreover, in the same study loss of Beclin1 function promotes gene amplification and chromosomal instability that result in aneuploidy, which are typical features of tumor. The author’s suggested that autophagy serves an important role in the preservation of genomic integrity. Furthermore, Karantza-Wadsworth et al., investigated the mechanism by which allelic loss beclin1 promotes breast tumorigenesis using mouse mammary epithelial cells (MMECs) and the author’s reported that allelic loss of beclin1 that resulted in defective autophagy sensitized mammary epithelial cells to metabolic stress and accelerated lumen formation in mammary acini. The author’s also reported that defective autophagy resulted in activation of DNA damage response in vitro and in mammary tumors in vivo that heightened gene amplification, and synergized with disabled apoptosis to accelerate mammary carcinogenesis.
Moreover, p62, a scaffolding protein involved in signal transduction events, as well as directing poly-ubiquitinated proteins and aggregates to autophagosomal degradation, serves as a critical link between defective autophagy and tumorigenesis. In this regard, in autophagycompromised cells, abnormal buildup of defective mitochondri a and protein aggregates could lead to accumulation in reactive oxygen species (ROS) that result in DNA damage along with p62 accumulation. Again p62 accumulation upon metabolic stress leads to ROS generation, thereby creating a positive feedback loop. It is therefore, autophagy contribute to oncosuppressive function by fighting against ROS accumulation and associated DNA damage.
Inhibition of Necrosis and Inflammation
Necrosis is a form of cellular demise characterized by several features such as ATP depletion, the loss of cellular osmolality, and release of various factors such as high mobility group box 1 protein (HMGB1) and breakdown of cell, which all lead to a strong inflammatory response. In this regard, tumor cells can evade this ATP-limiting demise by activating the energy sensor LKB1/AMPK complex, which, in turn, inhibits mTOR-C1, leading to activation of autophagy [14]. In this regard, activation of autophagy promotes cancer cells development in the face of inhibiting necrosis-associated inflammatory responses, because necrosis can promote the release of pro-metastatic immunemodulatory factors such as HMBG1, and this may lead to increased metastasis. Additionally, necrosis has been shown to be activated by AKT and Ras oncogenic signaling and is up-regulated when autophagy is compromised by mono-allelic deletion of BECN1. Thus, autophagy is perceived to avert necrosis in order to limit further cellular damage that may promote tumorigenesis and metastasis.
Promotion of Cancer Cells Death
In addition to promoting tumor cell survival at times of stress, autophagy could play in eliciting a type-II programmed cell death, known as autophagic cell death (ACD) [12]. ACD refers to cell death caused by autophagy rather than cell death with autophagy. Thus, the ultimate cell death process of ACD is executed by over-activated autophagic flux rather than apoptosis or necroptosis. The genetic or drug-based inhibitors of autophagy, but not apoptosis or necroptosis inhibitors rescue this type of cell death. Moreover, several studies have suggested a potential relationship between ACD and cancer progression. For example, one study showed that blocking the CXCR4/ mTOR signalling pathway induced ACD and the anti-metastatic properties of peritoneally disseminated gastric cancer cells [12]. The exact mechanism(s) connecting autophagy and apoptosis as partners in cell killing is still uncertain, however, apoptotic regulators antiapoptotic Bcl-2 family members Bcl-XL and Bcl-2 have been shown to control autophagy, and a cleaved form of Atg5 known to be autophagy regulating protein has been shown to induce apoptosis directly in the mitochondria [11,14].
Furthermore, co-activation of autophagy and apoptosis has also been shown in tumor cells. For instance, DRAM-1 was found to have both proautophagic and proapoptotic roles downstream from p53. However, a study done by Yee et al. reported that, p53 up-regulated modulator of apoptosis (PUMA) is the main mediator of p53-dependent apoptosis, and further the study revealed that, PUMA orchestrates this apoptosis by activating Bax and mitochondrial outer membrane permeabilization and release of cytochrome c. Moreover, the same study reported the capacity of PUMA to prompt autophagy that results in mitophagy. But, inhibition of PUMA or Bax-induced autophagy reduces the apoptotic response, suggesting the existence of synergy between autophagy and apoptosis in this apoptosis response. Although apoptosis and autophagy can occur in a cooperative manner to elicit cell death, these processes are sometimes mutually antagonistic [12]. For example, caspases (the effectors of the apoptotic cell death) have been shown to cleave and inactivate Atg6/Beclin 1, and the suppression of Atg6 function increases apoptotic cell death. Thus, apoptosis is either inhibited or delayed when autophagy is present, with the probable conclusion that autophagy is activated to protect cells from dying.
Other contrasting behaviors between apoptosis and autophagy have been attributed to the health status of the cell, or stage of transformation. Dominant-negative FADD invokes a death stimulus involving autophagy in healthy cells but not in cancer cells and induces varying amplitudes of death responses at different stages of cancer progression. In addition, oncogenic Ras has also recently been shown to cause ACD in the absence of apoptosis, even though other reports have indicated that autophagy is required for Ras-driven tumor growth. It is therefore, the decision to activate, repress, or simply not to manipulate autophagy, may be governed by the nature of the stimuli or the upstream regulator of an autophagic and/or apoptotic protein, as well as the health status of cells, and the net outcome of these three possibilities is either to promote or repress tumor survival, or both.
Maintenance of Normal Stem Cells
Autophagy appears to ensure maintenance of normal stem cells; this is particularly relevant for hematological malignancies, which are normally characterized by changes in proliferation or differentiation potential that alter the delicate equilibrium between toti-, pluri- and unipotent precursors in the bone marrow [4]. For instance, ablation of Atg7 in murine hematopoietic stem cells (HSCs) has been shown to disrupt tissue architecture, eventually resulting in the expansion of a population of bone marrow progenitor cells with neoplastic features. Similarly, tissue-specific deletion of the gene coding for FIP200 alters the fetal HSC compartment in mice, resulting in severe anemia and perinatal lethality. Interestingly, FIP200 gene knocked out murine HSCs do not exhibit increased rates of apoptosis, but an increased proliferative capacity. Moreover, Becn1+/_ mice display an expansion of progenitor-like mammary epithelial cells. Noteworthy, in human, autophagy has also known to be vital for conservation of normal stem cell compartments. Certainly, hematopoietic, dermal, and epidermal stem cells transfected with a short-hairpin RNA (shRNA) against ATG5 gene resulted in loss of self-renew ability while differentiating into neutrophils, fibroblasts, and keratinocytes, respectively.
Degradation of Oncogenic Proteins
Studies reported that autophagy is involved in the degradation of oncogenic proteins, including mutant (but not wild-type) TP53, p62, PML-RARA, and BCR-ABL1 [4,19]. It has been known that accumulation of mutant TP53 in tumor cells that functions as a principal-negative factor, has been recognized to abolish the antitumor role of the wild-type p53 [15]. Moreover, in a study done by Choudhury et al. reported that increased accumulation of mutant TP53 in tumor cells having low level of autophagy regulators ULK1, BECN1 or ATG5. But, the same study demonstrated that depletion of mutant TP53 in case of transgene-driven overexpression of Becn1 or ATG5. It is therefore, autophagy-dependent removal of mutant TP53 could reinstate the wild-type TP53 capacity of halting malignant transformation.
Moreover, p62 protein levels are frequently found to be up-regulated in human cancers and thought to promote tumorigenesis. According to studies, the accumulation of p62 leads to increased ER stress and DNA damage as well as it contributes to the deregulation of the nuclear factor kappa B (NF-kB) and antioxidant nuclear factor erythroid 2-related factor 2 (NRF2) pathways in cancer. Meanwhile, in the context of autophagy, p62 acts as an adaptor protein that links LC3 with ubiquitin moieties on misfolded proteins and degradation of p62 together with ubiquitylated proteins will be mediates by autophagy. At this juncture, defect in autophagic activity would cause accumulation of p62 and plays a role in carcinogenesis. In support of this notion, Takamura et al. reported that, mouse models with either a mosaic deletion of ATG5 or a liver-targeted deletion of ATG7 results in development of benign tumors with accumulation of p62. Furthermore, in the same study, deletion of p62 suppresses tumor growth, which indicates a causative link between p62 accumulation and adenoma formation.
Modulation of Anti-tumor Immune Response
It has been known that, antigens derived from outside the cell are subjected to degradation inside the lysosomes. In this regard, autophagy know to be involved in trafficking antigens meant for degradation, plus trafficking components from degraded antigen back to cell surface for presentation on the class II major histocompatibility complex (MHC). As reported by studies, most tumor antigens are derived from inside the cell, and there breakdown is mainly via proteasomes, leading to presentation on class I MHC. However, according to studies, autophagy stills plays a major role regarding to presentation of tumor antigens on antigen-presenting cells (APCs) directly on class II MHC or via a process termed “cross-presentation” on class I MHC. A study reported by Li et al. demonstrated that induction of autophagy by rapamycin in melanocytes significantly enhance the priming of APCs CD8+ T cells in presenting the melanocyte-derived tumor antigen gp100. However, in the same study, this effect has been reversed when autophagy is blocked using 3-methyladenine (3-MA, a PI3K III inhibitor). Subsequent study done by Lee et al. has established the the requirement of autophagy to enhance class I antigen presentation by dendritic cells (DCs). In this study the author’s reported that Atg5−/− DCs present antigen on class II molecules but has shown a reduced capacity to stimulate CD4+ lymphocytes via boosting antigen breakdown in autolysosomes. The same study also revealed that autophagy triggered by starvation and pharmacological treatment with rapamycin reduces class II presentation. As a result, both cancer cell-intrinsic and systemic defects in autophagy may prevent the host immune system to properly recognize and eliminate pre-malignant and malignant cells.
Contributes to Oncogene-induced Senescence
It has been established that irreversible form of cell cycle arrest could arise in response to stress signals, including telomere shortening and oncogene activation or oncogene-induced senescence (OIS) and it is known as cellular senescence [4]. Several studies have demonstrated that this phenomenon, which was initially identified and characterized in vitro, acts as a robust tumor suppressor mechanism in vivo. In this regard, it has been suggested that autophagy contributes to OIS. In support of this, Young et al. reported that several ATG genes, including ULK3, Atg7, and LC3 are up-regulated during oncogeneinduced senescence. Moreover, both pharmacological inhibitors of autophagy and small-interfering RNAs targeting ATG5, ATG7 or Becn1 prevented spontaneous senescence in primary human melanocytes or diploid fibroblasts while preventing the degradation of an endogenous, dominant-negative TP53 variant [4]. Meanwhile, much thought has been put into the precise role of autophagy in OIS. White and Lowe reported that autophagy is evoked to break down specific cellular components to enable physical remodeling associated with senescence.
Antiviral and Antibacterial Effects
Autophagy may suppress carcinogenesis owing to its key role in the first line of defense against viral and bacterial infection. Actually, autophagy has been stimulated during infection by numerous carcinogenic pathogens. For instance, viruses including, hepatitis B virus, human papillomavirus (HPV), Epstein–Barr virus have been associated with the pathogenesis of HCC, cervical and gastric carcinoma, respectively. On the other hand, bacterial pathogens including, Streptococcus bovis, Chlamydia pneumonia and Helicobacter pylori have been associated with colorectal, lung and gastric carcinoma, respectively. In addition, Salmonella enterica has been associated with an increased incidence of Crohn’s disease, hence sustaining colorectal carcinogenesis, and gallbladder carcinoma.
This selective autophagy induced by and against pathogens known as xenophagy [3]. According to studies, xenophagic response is required for the rapid clearance of intracellular pathogens as well as for the stimulation of pathogen-specific immune responses. Consequently, reduced levels of autophagic markers including Becn1 have been correlated with HPV-16 and HPV-18 infection in a cohort of cervical carcinoma patients. Therefore, autophagy may exert oncosuppressive effects also by virtue of its antiviral and antibacterial activity.
Promotion of Tumor Progression
Despite its role in preventing early tumor development, once tumors are established, tumor cell autophagy-related survival function can lead to tumor dormancy, progression, and therapeutic resistance. Furthermore, advanced human cancers exhibited an amplified autophagic flux, linked with invasive phenotype, high nuclear grade, and poor disease outcome. Proposed mechanisms for tumor-supporting functions of autophagy in established tumors includes resistance to stress induced by hypoxia, starvation and cancer therapy, survival of senescent cancer cells, maintenance of cancer stem cells (CSCs), resistance to epithelial-to-mesenchymal transition (EMT) [4].
Supports Survival of Tumor Cells Under a Variety of Stresses
In established tumor, metabolic stress known to induce autophagy so as to seek an alternative source of energy and metabolites. Furthermore, autophagy may also be induced as an adaptive cellular response to various cancer therapies, leading to chemo-resistance and cancer cell survival.
As shown in Table 2, inhibition of autophagy through treatment with 3-MA, bafilomycin A (a specific inhibitor of vacuolar-type H+-ATPase), chloroquine (CQ) or hydroxyl-chloroquine (HCQ) {lysosome-tropic agents that impair fusion between autophagosomes and lysosomes}, or silencing of some essential autophagy genes, such as ATG3, ATG4B, ATG5, Atg6/BECN1, ATG7, ATG10, and ATG12 has been found to increase cell death to a wide spectrum of therapeutic stresses. Moreover, activation of autophagy has been considered as one mechanism for acquired resistance of tumor cells to chemotherapeutic agents.
| Agent | Class | Tumor type | Role of autophagy |
|---|---|---|---|
| Rapamycin | mTORC1 inhibitors | Malignant glioma | pro-death |
| Everolimus | Acute lymphoblastic leukemia, prostate cancer | pro-death | |
| Temsirolimus | Mantle cell lymphoma | pro-death | |
| AZD8055 | Lung cancer | pro-death | |
| Imatinib | Tyrosine kinase inhibitors | Chronic myeloid leukemia | pro-death |
| INNO-406 | Gastrointestinal stromal tumor | pro-survival | |
| Chronic myeloid leukemia | pro-survival | ||
| Dasatinib | Ovarian cancer | pro-death | |
| Chronic lymphocytic leukemia | pro-survival | ||
| Perifosine | Akt inhibitors | Chronic myelogenous leukemia | pro-survival |
| Triciribine | Acute lymphoblastic leukemia | pro-survival | |
| Metformin | AMPK activators | Cervical cancer | unknown |
| Melanoma | pro-death | ||
| Bortezomib | Proteasome inhibitors | Cervical cancer | pro-death |
| Prostate cancer | pro-survival | ||
| NPI-0052 | Prostate cancer | pro-survival | |
| Vorinostat | Histone deacetylase (HDAC) inhibitors | Gastric cancer | pro-survival |
| SAHA | Chondrosarcoma, endometrial stromal sarcoma, HCC | pro-death | |
| MHY218 | Breast cancer | pro-death | |
| S1 | Bcl-2 inhibitors | Malignant glioma | pro-survival |
| Z18 | Breast cancer | pro-death pro-death |
|
| (–)-gossypol | prostate cancer | ||
| Tamoxifen | Estrogen receptor | Breast cancer | pro-survival |
| modulator | |||
| Nelfinavir | HIV-protease inhibitor | Multiple cancer | pro-death |
| Etoposide | Topoisomerase II inhibitor | HCC | pro-death |
| 3-MA | PI3K inhibitor | Colon cancer, Prostate cancer | pro-survival |
| CQ | Lysosomotropic agents | Glioblastoma, colon cancer | pro-survival |
| HCQ | Breast cancer | pro-survival | |
| Bafilomycin A1 | Vacuolar ATPase inhibitor | Breast cancer, colon cancer | pro-survival |
Table 2: Representative autophagy-modulating agents in cancer therapy.
Sustains Survival of Senescent Cancer Cells
Tumor dormancy, characterized by asymptomatic nature of tumor for extended periods of time, can be present as one of the earliest stages in tumor development, as well as in the in micro-metastasis stage, and can occur when minimal residual disease remains after surgical removal or treatment of primary tumors. It can result from angiogenesis arrest, in balance between apoptosis and cell proliferation, cell cycle arrest, immune surveillance and cancer therapy. It is a poorly understood phase of cancer progression and only recently have its underlying molecular mechanisms started to be revealed. In a study done by Lu et al. revealed that, when the level of ARHI remains at normal autophagic activity, tumor dormancy is likely to occur, as removed tumors can recover their abilities that are consistent with the autophagic process. In the study, the inhibition of ARHI-induced autophagy can dramatically reduce the regrowth of xenografted tumors upon the reduction of ARHI levels, suggesting that autophagy may contribute to the survival of dormant cells; therefore, ARHI can induce ACD as well as promoting tumor dormancy in the presence of factors that promote survival in the tumor microenvironment.
Furthermore, cancer cells exposed to therapeutic interventions can also undergo senescence. In this regard, senescent cells do not proliferate, but they may support disease relapse by releasing a wide panel of proinflammatory and mitogenic cytokines into the microenvironment the so-called senescence-associated secretory phenotype (SASP). However, according to studies, these cells are greatly reliant on autophagy for survival, and inhibitions of autophagy have been shown to synergize with several cancer therapeutics in experimental models of lymphoma that are susceptible to acquire the SASP in response to treatment.
Ensures the Maintenance of the Cancer Stem Cell Compartment
It has been known that, autophagy has played a role in the development of resistance to cancer therapy, self-renewal, differentiation and tumorigenic abilities of CSCs. A study done by Song et al. showed that CD133+ liver CSCs (LCSCs) significantly enrichment with higher autophagy levels after hypoxia and nutrient starvation in HCC Huh 7 cells. It is therefore, the study reported the autophagy’s essential role in LCSC maintenance. Furthermore, conversion of microtubuleassociated protein LC3-I to LC3-II as well as improved buildups of ATG7 and Beclin-1 has been observed in pancreatic CSCs treated with Rottlerin (ROT), which has been commonly used protein kinase C-delta (PKC-δ) inhibitor. Additionally, the gene silencing of ATG7 and Beclin-1, or co-treatment with 3-MA, can inhibit ROT-induced ACD.
In addition, expressions of ATG5, ATG12 and LC3B in dormant stem cell-like breast cancer cells by farnesyl transferase inhibitors (FTIs) in breast cancer cells having low metastatic abilities induced a reversible state of dormancy. Moreover, JNK-mediated autophagic pathway was also up-regulated in these breast CSCs with the periods of FTIinduced dormancy. On the other hand, irradiation of CD133+ glioma stem cells (GSCs) has been associated with induction of autophagy in a short time and then autophagy slightly decreases the viability of the cells. According to the study, the gamma-radiation also induced a large degree of autophagy in the CD133+ GSCs, characterized by high level expression of ATG proteins such as ATG5, LC3 and ATG12.
Modulation of Autophagy in Cancer Therapy
The role and regulation of autophagy in cancer is apparently quite complex, but when tumor cells induce protective autophagy, inhibition of autophagy may provide a way of sensitizing tumor cells to therapy by activating apoptosis. Conversely, excessive autophagy induced by drugs can potentially result in tumor cell elimination [12]. On top of this, role of autophagy as a protector of cellular homeostasis and genome integrity may be particularly important regarding cancer prevention.
Induction of Autophagic Cell Death (ACD)
Defects in apoptosis are often associated in many cancer cells; hence targeting alternative cell death pathways is an attractive strategy for improving anti-tumor therapy. Thus, the induction of cell death by autophagy may serve as a novel therapeutic strategy, especially in cancer cells with high thresholds to apoptosis.
Moreover, the consequence of promoting autophagy depends on multiple factors, including extent of induction, duration, and cellular context. Accumulating evidence indicates that promotion of autophagy that leads to ACD contributes to in vivo antitumor effects (Table 2). For instance, a natural BH3-mimetic, small-molecule inhibitor of Bcl2, (-)-gossypol, shows potent antitumor activity against human prostate cancer. Accordingly, the antitumor activity by (-)-gossypol is mediated through induction of both apoptosis and ACD. However, autophagy appears to serve as a death program primarily when the apoptotic machinery is defective, as observed in most tumors.
Inhibition of Protective Autophagy
Many anti-cancer therapies such as DNA damaging agents, radiation therapy and targeted therapies can induce autophagy in cancer cells. According to studies, cytotoxic chemotherapeutics and targeted therapies induce autophagy through a number of signaling pathways including the DNA damage response, mTOR and AMPK signaling, the ER stress response and others. In this regard, autophagy is commonly up-regulated in both tumor and normal cells exposed to cancer therapies, but the greater reliance of tumor cells (versus normal cells) on the cytoprotective effects of autophagy and its potential clinical relevance in modulating drug resistance. Consistence with this notion, several studies have found that inhibition of chemotherapy-induced autophagy can enhance the sensitivity of cancer cells to different chemotherapies, leading to cell death and tumor regression.
For instance, the apoptosis potential of melanoma differentiationassociated gene-7/Interleukin-24 (MDA-7/IL-24), a member of IL- 10 gene family that has been shown a nearly ubiquitous antitumor properties in vitro and in vivo through induction of cancer-specific apoptosis, increases by inhibiting protective autophagy with 3-MA (Table 2) in prostate cancer cells. Similarly, inhibition of autophagy by 3-MA or Atg7 knockdown induced apoptosis in colon cancer cells treated with 5-FU. Moreover, inhibition of protective autophagy was shown to sensitize resistant cells to TRAIL-mediated apoptosis in apoptosis-defective leukemic and colon cancer cell lines (Table 2). In this regard, these apoptosis-resistant cells only become apoptotic after inhibition of autophagy.
Furthermore, the potential to inhibit autophagy and sensitize tumor cells to metabolic stress is another promising approach for cancer therapy. Many current cancer therapies including angiogenesis, growth factor, and receptor inhibitors when combined with autophagy inhibition produced synergistic anticancer effects. Moreover, combining organelle-damaging drugs, such as sigma-2 receptor agonists, with an autophagy inhibitor might be an effective means of cancer therapy. It is likely that ER stress inducers, including thapsigargin and tunicamycin, that trigger cell death in cancer cells will increase cell killing when autophagy is inhibited. On the other hand, targeting both proteasomeand autophagy-mediated protein degradations might be an effective antitumor approach for highly metabolically active tumor cells. The proteasome inhibitor bortezomib has the approval of the US FDA and has demonstrated potent efficiency in treating multiple myeloma.
Moreover, metastasis prone state of cancer cells may be principally subject to autophagy inhibition as cells in isolation are likely to be more dependent autophagy, even though this likelihood remains to be established. Incidentally, CQ that has been known to inhibit lysosome by acidification and thereby inhibition of autophagy, in conjunction with alkylating agents, displayed remarkable efficacy in inhibiting tumor growth in mice as well as in clinical studies. Synergy between CQ and the HDAC inhibitor suberoylanilide hydroxamic acid (SAHA) in killing imatinib refractory chronic myeloid leukemia cells also supports a protective role for autophagy, reinforcing the therapeutic use of autophagy inhibitors in cancer therapy [6]. Similarly, the synergy between CQ and the PI3K/mTOR inhibitor NVP-BEZ235 induced apoptosis in glioma xenografts. By the same token, cyclophosphamideinduced tumor cell death in a Myc-induced murine lymphoma enhanced by CQ in a similar fashion produced by shRNA knock down of Atg5, and it hindered the time-to-tumor recurred. These studies documented that CQ, or its analog (HCQ), when used as autophagy inhibitors in combination with proapoptotic drugs, increases twofold the median survival of cancer patients.
Currently, multiple clinical trials are assessing the effects of combined treatments with various anti-cancer drugs plus HCQ for patients with various refractory malignancies (Table 3). Meanwhile, one underlying concern is that autophagy inhibitors approved for cancer patients might actually act as promoters of tumor development. However, the tumorpromoting effect of autophagy inhibitors, which depends on necrotic cell lysis that follows the inflammatory response, could prevent this undesirable effect upon co-treatment with immunosuppressive drugs.
| Cancer type | Clinical trials | Drug (s) | Phase |
|---|---|---|---|
| Advanced solid tumors | NCT00909831 | HCQ, temsirolimus I | I |
| NCT01266057 | HCQ, sirolimus, vorinostat | I | |
| NCT01023737 | HCQ, vorinostat | I | |
| NCT01480154 | HCQ, MK2206 | I | |
| Breast cancer | NCT01292408 | HCQ | II |
| Colorectal cancer | NCT01206530 | HCQ, leucovorin, 5-FU, oxaliplatin, bevacizumab | I/II |
| NCT01006369 | HCQ, capecitabine, oxaliplatin, bevacizumab | II | |
| Ductal carcinoma in situ | NCT01023477 | CQ | I/II |
| Epithelial ovarian cancer | NCT01634893 | HCQ, sorafenib | I |
| Extraovarian peritoneal carcinoma | NCT01634893 | HCQ, sorafenib | I |
| Fallopian tube carcinoma | NCT01634893 | HCQ, sorafenib | I |
| Kidney cancer | NCT01480154 | HCQ, MK2206 | I |
| Lymphangioleiomyomatosis | NCT01687179 | HCQ, sirolimus | I |
| Melanoma | NCT00962845 | HCQ | 0 |
| Multiple myeloma | NCT00568880 | HCQ, bortezomib | I/II |
| NCT01438177 | CQ, velcade, cyclophosphamide | II | |
| Non-small cell lung cancer | NCT00933803 | HCQ, carboplatin, paclitaxel, bevacizumab | I/II |
| NCT01649947 | HCQ, carboplatin, paclitaxel, bevacizumab | II | |
| Pancreatic cancer | NCT01506973 | HCQ, gemcitabine | I/II |
| NCT01128296 | HCQ, gemcitabine | I/II | |
| Prostate cancer | NCT01480154 | HCQ, MK2206 | I |
| Renal cell carcinoma | NCT01144169 | HCQ | I |
| NCT01510119 | HCQ, RAD001 | I/II | |
| Small cell lung cancer | NCT00969306 I | CQ, A-CQ 100 | I |
Table 3: Ongoing clinical trials evaluating the efficiency of CQ/HCQ anticancer activity.
Future Perspectives
There are still several questions that remains to be answered even though, incredible amount of progress have been achieved about role of autophagy in cancer. For instance, the switch in regulation of autophagy from low levels, i.e. during tumorigenesis to elevated levels during tumor progression, has not been clarified. Moreover, the delineation of the autophagic cargo in tumors with elevated autophagy will allow for a greater understanding of how this process is integrated into the metabolism of the cell. Most likely, there will be differences in the role of autophagy in normal metabolism versus that of its tumorigenic counterpart. Given the progress that has been made in proteomic and metabolomics approaches, this type of analysis is certainly feasible.
References
- Ktistakis NT, Tooze SA (2016) Digesting the Expanding Mechanisms of Autophagy. Tr Cell Biol 26: 624-635.
- Mancias JD, Kimmelman AC (2016) Mechanisms of selective autophagy in normal physiology and cancer. J Mol Biol 428: 1659-1680.
- Zaffagnini G, Martens S (2016) Mechanisms of Selective Autophagy. J Mol Biol 428: 1714-1724.
- Galluzzi L, Pietrocola F, Pedro J, Amaravadi R, Baehrecke E, et al. (2015) Autophagy in malignant transformation and cancer progression. EMBO J 34: 856-880.
- Gallagher L, Williamson L, Chan E (2016) Advances in Autophagy Regulatory Mechanisms. Cells 5: 24.
- Papinski D, Kraft C (2016) Regulation of Autophagy by Signaling Through the Atg1/ULK1 Complex. J Mol Biol 428: 1725-1741.
- Park J, Jung C, Seoa M, Otto N, Grunwald D, et al. (2016) The ULK1 complex mediates MTORC1 signaling to the autophagy initiation machinery via binding and phosphorylating ATG14. Autophagy 12: 547-564.
- Kruppa AJ, Kendrick-Jones J, Buss F. Myosins S (2016) Actin and Autophagy. Traffic 17: 878-890.
- Kim Y, Park J, Grunwald D, Kim D (2016) An expanded role for mTORC1 in autophagy. Mol Cellular Oncol 3: e1010958.
- Sridharan S, Jain K, Basu A (2011) Regulation of Autophagy by Kinases. Cancers 3: 2630-2654.
- Antonioli M, Di Rienzo M, Piacentini M, Fimia GM (2016) Emerging Mechanisms in Initiating and Terminating Autophagy. Trends Biochem Sci 968: 30171-30182.
- Liang C (2010) Negative regulation of autophagy. Cell Death and Differentiation 17: 1807-1815.
- Jeon Sang-Min (2016) Regulation and function of AMPK in physiology and diseases. Exper Mol Medi 48: e245.
- Kim J, Kim YC, Fang C, Russell RC, Kim JH, et al. (2012) Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell. 152: 290-303.
- Chandra V, Bhagyaraj E, Parkesh R, Gupta P (2008) Transcription factors and cognate signalling cascades in the regulation of autophagy 91: 429-451.
- Kim J, Kundu M, Viollet B, Guan KL (2006) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13: 132-141.
- Dorsey Waneene C (2015) Mitogen-Activated Protein Kinase (MAPK) Signaling Pathway and Mammalian Cells. Transcriptomics 3: 2.
- Tang JC, Feng YL, Liang X, Cai XJ (2016) Autophagy in 5-Fluorouracil Therapy in Gastrointestinal Cancer: Trends and Challenges. Chin Med J 129: 456-463.
- Ruocco N, Costantini S, Costantini M (2015) Blue-Print Autophagy: Potential for Cancer Treatment. Mar Drugs 14: 138.
- Maycotte P, Thorburn A (2014) Autophagy and cancer therapy. Cancer Biol Ther 11: 127-137.
- Gunn JM, Clark MG, Knowles SE, Hopgood MF, Ballard FJ (1977) Reduced rates of proteolysis in transformed cells. Nature 266: 58-60.
- Kisen GO, Tessitore L, Costelli P, Gordon PB, Schwarze PE, et al. (2002) Reduced autophagic activity in primary rat hepatocellular carcinoma and ascites hepatoma cells. Carcinogenesis. 14: 2501-2505.
- Aita VM, Liang XH, Murty VV, Pincus DL, Yu W, et al. (1999) Cloning and genomic organization of beclin 1, a candidate tumor suppressor gene on chromosome 17q21. Genomics59: 59-65.
- Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, et al. (2005) Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 402: 672-676.
- Yue Z, Jin S, Yang C, Levine AJ, Heintz N, et al. (2003) An autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. P Natl Acad Sci 100: 15077-15082.
- Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, et al. (2002) Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest 112: 1809-1820.
- Deng Q, Wang Z, Wang L, Zhang L, Xiang X, et al. (2013) Lower mRNA and protein expression levels of LC3 and Beclin1, markers of autophagy, were correlated with progression of renal clear cell carcinoma. Jpn J Clin Oncol 43: 1261-1268.
- Rothe K, Lin H, Lin KB, Leung A, Wang HM, et al. (2014) The core autophagy protein ATG4B is a potential biomarker and therapeutic target in CML stem/progenitor cells. Blood 123: 3622-3634.
- Liang C, Feng P, Ku B, Dotan I, Canaani D, et al. (2006) Autophagic and tumor suppressor activity of a novel Beclin1-binding protein UVRAG. Nat Cell Biol 8: 688-699.
- Kim MS, Jeong EG, Ahn CH, Kim SS, Lee SH, et al. (2008) Frameshift mutation of UVRAG, an autophagy-related gene, in gastric carcinomas with microsatellite instability. Hum Pathol 39: 1059-1063.
- Tang J, Deng R, Luo RZ, Shen GP, Cai MY, Du ZM, et al. (2012) Low expression of ULK1 is associated with operable breast cancer progression and is an adverse prognostic marker of survival for patients. Breast Cancer Res Treat 134: 549-560.
Share This Article
Recommended Journals
Open Access Journals
Article Tools
Article Usage
- Total views: 6380
- [From(publication date): 0-2018 - Feb 04, 2025]
- Breakdown by view type
- HTML page views: 5631
- PDF downloads: 749