Research Article Open Access
Arsenic Occurrence and Fate in the Environment; A Geochemical Perspective
| Panagiotaras D1 and Nikolopoulos D2* | |
| 1Department of Mechanical Engineering, Technological Educational Institute (TEI) of Western Greece, M. Alexandrou 1, 263 34 Patras, Greece | |
| 2Department of Electronic Computer Systems Engineering, Piraeus University of Applied Sciences, Petrou Ralli and Thivon 250, GR 122 44, Aigaleo, Athens, Greece | |
| Corresponding Author : | Nikolopoulos D Department of Electronic Computer Systems Engineering Piraeus University of Applied Sciences (TEI of Piraeus) Petrou Ralli and Thivon 250, 122 44 Aigaleo, Greece Tel: +0030-2105381560 E-mail: dniko@teipir.gr |
| Received February 15, 2015; Accepted March 18, 2015; Published March 28, 2015 | |
| Citation: Panagiotaras D, Nikolopoulos D (2015) Arsenic Occurrence and Fate in the Environment; A Geochemical Perspective. J Earth Sci Clim Change 6:269. doi: 10.4172/2157-7617.1000269 | |
| Copyright: © 2015 Panagiotaras D, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. | |
Visit for more related articles at Journal of Earth Science & Climatic Change
Abstract
The arsenic minerals in the environment constitute the primary source of the environmentally occurring arsenic. The As minerals interact with the environment and this renders either their dissolution or the formation of secondary minerals, or both. The distribution of the environmental arsenic is determined by the biogeochemical transformations with respect to the redox conditions, the pH, the availability of ions, the adsorption-desorption, dissolution and the biological activity. The arsenic in the environment is sorbed primarily by metal oxides, especially the ones of iron (Fe), aluminum (Al) and manganese (Mn). These are thought to bind As (+5) readily than As (+3). The overall adsorption depends greatly on pH. Metal oxides such as the ones of hydrous ferric, manganese and aluminum are additional important sinks of arsenic that is adsorbed or co-precipitated. Their dissolution depends also on pH. The redox potential and the microbial activity. Final result is the release of arsenic chemical species into the environment. This review presents a systematic compilation of the major geochemical processes that govern arsenic fate in the environment. The paper attempts to compile the removal capacity of constituents which could be useful for the purpose of As remediation.
| Keywords |
| Arsenic environmental fate; Arsenic speciation; Arsenic geochemistry |
| Introduction |
| Arsenic (As) is a trace inorganic element. It is found in the crystal lattice of arsenic minerals inspected in the environment. It may also arise from arsenic traces adsorbed onto the surfaces of -other minerals as inner or outer sphere complexes. Arsenic minerals–water interaction govern the arsenic occurrence in water systems. Due to this, arsenic is traced in surface and ground waters, -for example in rivers, stream, sea waters and lakes. The rate of interaction between water and arsenic minerals depends on several ad-hoc biogeochemical conditions such as the redox potential (Eh), the pH, the temperature, the microbial activity, the speciation and the concentration of metals in the fluid and the ionic strength of the solution. Once arsenic is released it can be sorbed onto the minerals phase, precipitate, redissolved and biointegrate according to the surrounding environmental conditions. |
| Arsenic pollution has become a significant environmental problem nowadays. Arsenic is a contaminant of concern regarding many industrial products, wastes and wastewaters and is widely known for its adverse effects on human health. Great human populations have been affected by the adverse health effects of arsenic [1,2]. Recent epidemiological studies have reported noteworthy detrimental effects of As on humans due to its high toxicity. The toxicity occurs even at the ppb level [1,2]. Due to the above reasons, the World Health Organization (WHO) suggested that the arsenic concentration in drinking water should not exceed 10 μg/L [1]. Note, that the upper concentration limit of As in drinking water was 50 μg/L [1]. Arsenic is a known neurotoxin [2]. There are several studies which link arsenic exposure to neuropathological disorders such as production of ß amyloid [3], hyperphosphorylation of tau protein [4], oxidative stress [5], inflammation [6], endothelial cell dysfunction [7], and angiogenesis [8]. Note that some of these disorders are related to cognitive dysfunction and Alzheimer’s disease [9]. Morphologic and neurochemical alterations have also been observed in animals during arsenic exposure. These have been associated with the hippocampus and other memory-related neuronal structures related learning and memory [2,7]. Skin diseases and carcinogenesis are other effects observed due to the arsenic exposure according to human epidemiological studies [10]. Most importantly, considerably high were the estimated cancer risks for wide population groups which were exposed to arsenic-contaminated water in Taiwan, Japan, Bangladesh, West Bengal-India, Chile and Argentina [11]. The higher risks were identified for cancer of the skin and then for cancers of lung, bladder, kidney and liver [11]. |
| Nowadays several soil and groundwater reservoirs have been contaminated with arsenic. This has created the need for remedy against arsenic contamination. However, the treatment and remediation of As is not an easy task to accomplish since arsenic changes several valence states and reacts towards the formation of species with varying toxicity and mobility. It is this reason that makes the removal of arsenic from drinking water a really recurring challenge, especially in the developing countries. It should be noted that there are several reasons apart from cost that can make it advantageous to use local materials as adsorbents for arsenic. Such materials can be retrieved from the nearby soils as well as the topical agriculture and industry. |
| Of course these materials are not always optimal, but their availability at various locations all over the world often makes them attractive choices. |
| Arsenic Origin in the Environment |
| Traces of arsenic can be found in several parts of the natural environment. Arsenic is traced in air, soil, water, rocks, plants and animals. There are various natural activities that release arsenic to the environment. Most significant are the volcanic eruption, the erosion of the rocks and the forest fires. Arsenic is widely distributed in more than 320 minerals [12]. The most common arsenic minerals are: |
| (1) arsenopyrite (FeAsS) [13-15]; |
| (2) orpiment (As2S3); |
| (3) realgar (As2S2); |
| (4) pyrite (FeS2) where arsenic is found as a solid solution [16,17]. |
| After weathering of arsenic minerals the arsenic ions can be adsorbed onto the Fe (III) and Mn (IV) oxide-hydroxide phases [15,18-20]. The arsenic bearing minerals are also significant sources of arsenic present in natural water. Apart from that, traces of As in natural water are observed due to the widespread use of [arsenic in pigments, insecticides and herbicides. Note that approximately 70% of the arsenic used, is in pesticides. In the form of pesticides it is principally traced in [21]: |
| (1) Monosodium methane arsenate (MSMA) – HAsO3CH3Na; |
| (2) Disodium methane arsenate (DSMA) – Na2AsO3CH3 and |
| (3) Dimethylarsinic acid (cacodylic acid) – (CH3)2AsO2H; |
| (4) Arsenic acid – H3AsO4 |
| In general, variable and low is the concentration of the arsenic in the environment [22]. For example, the average concentration in air in remote and rural areas ranges between 0.02 ng.m-3 and 4 ng.m-3 [22]. In urban areas the ambient concentration of arsenic is from 3 ng.m-3 to about 200 ng.m-3. High values, above 1000 ng.m-3, can be measured near industries. In ocean waters arsenic concentrates typically between 1 μg.l-1 and 2 μg.l [22]. Arsenic is also widely distributed in surface fresh waters, rivers and lakes. Note that concentrations below 10 μg.l-1 are usually addressed in rivers and lakes, however certain water samples may present arsenic concentration up to 5 mg.l-1, especially near anthropogenic sources [22]. The levels of arsenic in groundwater average to about 1-2 μg.l-1. Worth to mention is that in areas with volcanic rock and sulfide mineral deposits, the arsenic levels may rise up to 3 mg l-1 [22]. |
| There are numerous regions in the planet where the arsenic concentrations exceed the newly suggested Maximum Contaminant Level (MCL=10 μg.l-1) [1]. For example, waters in U.S., India and China, exceeds MCL value. It is notable that in Bangladesh about 30% of the ground water sources, have arsenic concentrations above the MCL concentration limit, exceeds 50 μg.l-1 [1]. In U.S., approximately 10% of the measured borehole water samples exceed 10 μg.l-1 [23]. Extremely high arsenic concentrations have been reported in water samples of Xinjiang (China), where the corresponding arsenic concentrations were all well above 50 μg.l-1 and more specifically between 50 μg.l-1 and 1860 μg.l-1 [24]. Another disconcerting issue is that high arsenic bioaccumulation rates in the dry weight range of 0.007 μg.g-1 and 125.9 μg.g-1 have been reported, as a result of its gathering into the tissues of aquatic biota [25]. Table 1 presents the major arsenic sources in earth materials. |
| The concentration of arsenic in sedimentary materials range from 1.7 to 400 mg.kg-1 [26]. Higher levels occur in contaminated areas. The background concentration of arsenic in soil is between 1 mg.kg-1 and 40 mg.kg-1, with a mean value around 5 mg.kg-1 [27]. Significant is that near industries, mines and mine tailings, the arsenic concentration in the soil and soil sediments is much higher [1]. For example, an average arsenic concentration of 903 mg.kg-1 was detected in mine tailings in British Columbia [28]. On the other hand, the marine organisms contain arsenic residues typically from 1 mg.kg-1 to about 100 mg.kg-1. Predominantly higher arsenic concentrations are addressed in organic arsenic species such as the arsenosugars (macroalgae) and the arsenobetaine (invertebrates and fish) [22]. Various anthropogenic activities act as additional sources of arsenic in the environment as for example farming, mining, uses of fossil fuels, pulp and paper production and cement manufacturing [29]. Other significant anthropogenic activities contributes to an input of Table 1-arsenic and its compounds in the environment are wood preservatives, glass manufacture, electronics, catalysts, alloys, feed additives and veterinary chemicals. |
| Fate of Arsenic in Nature |
| Since the primary source of arsenic in the natural environment is the arsenic minerals, the stability of these is a significant factor of controlling arsenic occurrence in nature. The interaction of the arsenic minerals with the environment results either to their dissolution, or to the formation of secondary arsenic minerals, or even both. Table 2, presents characteristic cases of solubility data of representative arsenic minerals and the Gibbs free energy of formation in their standard reference state (fG0) [reference 30 and references therein]. Note that the values of pH and the corresponding total arsenic solubility of Table 2, are the highest and lowest levels presented in the literature (Table 2). |
| The most common valence states of arsenic in natural systems are the +3 and +5 states. However, arsenic can be found in the (-3) oxidation state (arsine), and an arsenic compound in this state is extremely toxic. This toxic oxidation state can be formed under very reducing conditions, and for this reason its occurrence in nature is relatively rare. On the other hand, inorganic and organic species of As are present in the natural environment, with inorganic forms been typically more abundant in freshwater systems. |
| In aqueous systems, arsenic exhibits anionic behaviour. In case of oxygenated waters, arsenic acid predominates only at extremely low pH values, namely for pH below 2. In the pH range of 2 to 11, it is in the form of H2AsO4– and HAsO42-. In mildly reduced conditions and low pH values, arsenious acid is converted to H2AsO3– and this conversion is more frequent as pH increases. When the pH exceeds 12 HAsO32- does appear (Figure 1). |
| Arsenate [As(+5); HxAsO4x-3, x=0-3] and arsenite [As(+3);HxAsO3x-3, x=0-3] are the two most common inorganic forms of arsenic in freshwaters. As(+5) is thermodynamically stable under oxic conditions, while As(+3) is stable under more reducing conditions. However, As(+5) and As(+3) are often found in both oxic and anoxic waters and sediments. The oxidation of As(+3) by O2 is slow (on the order of several weeks), while bacterially-mediated redox reactions can be much faster [30-33]. |
| Arsenate is an anion at the pH of most natural waters (H2AsO4- and HAsO42-), while arsenite is a neutral specie. The pKa values for arsenate (H2AsO4) are pKa1=2.19, pKa2=6.94 and pKa3=11.5 according to the following equations (1-4): |
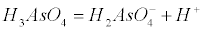 (1) (1) |
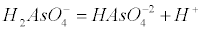 (2) (2) |
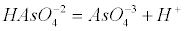 (3) (3) |
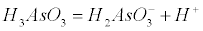 (4) (4) |
| In oxidative environments the form H2AsO4- predominates with pH values below 6.9, whereas the HAsO4-2 ions predominate at higher pH levels. Arsenite (H3AsO3) pKa value according to equation (4) is equal to 9.22, while it is the main arsenic chemical specie in natural waters with pH<9 and in slightly reducing conditions [34]. |
| In aerobic waters, arsenic acid predominates only at extremely low pH (<2). At the pH range of 2 to 11, it is replaced by H2AsO4- and HAsO42- ions. Arsenious acid appears at low pH and under mildly reduced conditions, but it is replaced by H2AsO3- as the pH increases. Only when the pH exceeds 12 the HAsO32- ion appears (Figure 1). The HAsS2 arsenic chemical specie can form at low pH in the presence of sulphide ions. Arsine derivatives and arsenic metal can occur under extreme reducing conditions [35]. |
| Arsenic changes its valence state and chemical form in the environment. In the pH range of 4 to 10, As(+5) species are negatively charged in water, and the predominant As(+3) species is neutral in charge (Figure 1). |
| The pH, the redox potential and the arsenic speciation are the critical parameters for the Eh-pH diagrams as those of Figure 1. If the half-reaction for the reduction of As(+5) to As(+3) is accounted in equation (5), the Nernst equation (6) relates the arsenic species concentrations at equilibrium [36], namely: |
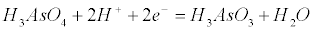 (5) (5) |
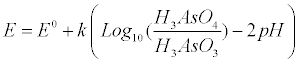 (6) (6) |
| In equation 6, E0 and k are a constant and a collection of constants, respectively. The dependence of the As(+5) fraction with the pH of the studied aquatic system is given by equation (7) as proposed by Stumm and Morgan 1996 [37]: |
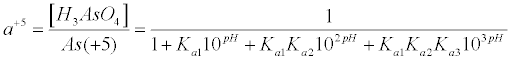 (7) (7) |
| A similar equation can be derived for the fraction of As(+3) in the H3AsO3 form (α+3) due to equation (8): |
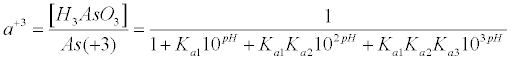 (8) (8) |
| Substitution of equations (7) and (8) into equation (6) give equation (9), which relates the equilibrium redox potential to the pH and concentrations of As(+5) and As(+3) measured quantities: |
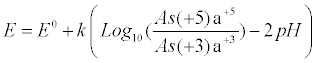 (9) (9) |
| The major conditions responsible for different arsenic valance and chemical species are redox potential, the presence of complexing ions, such as ions of sulfur, iron, calcium and microbial activity. Since arsenic forms anions in solution, it does not form complexes with simple anions like Cl– and SO42– as do cationic metals, whereas anionic arsenic complexes behave like ligands in water. |
| Arsenic Geochemistry |
| The geochemical processes which are involved in the oxidation, reduction and bioavailability of chemical species of arsenic play a significant role in nature [34]. Arsenic has been associated to microorganisms as well and especially their metabolism [34]. Arsenic is partitioned between solid and dissolved phases and this is associated with many biogeochemical processes. There are also several reactions that control the mobilization of arsenic in the natural environment. Most significant are the processes of dissolution-precipitation, adsorption-co precipitation and reduction-oxidation. |
| Oxidizing arsenic bacteria, oxidize As(+3) enzymatically and produce arsenite oxidases. Oxygen and nitrate reducing microorganisms, utilize arsenite as an electron donor. Characteristic examples are the photoautotrophic, heterotrophic and chemoautotrophic microorganisms. In these micro-organisms, the adduced energy is used for the production of CO2 which is required for the generation of carbon and the growth of bacteria community [34,38]. The oxidation of the As(+3) species to the less bioavailable As(+5) compounds is crucial for the detoxification processes [34,38]. Such processes can be observed in extreme natural environments [34,38]. They are considered as primary energy resources for the chemolithotrophic metabolism of organisms in the era of the formation of the very first forms of life [34,38]. The microorganisms which can act as oxidizers of arsenite, can also facilitate As(+3) oxidation in aerobic environments [37], viz. in the presence of O2. They can also act as electron acceptors in anoxic conditions by using other ions in the order of NO3-→Mn oxides, →Fe(III) oxides, →sulfate [37]. On the contrary, in the case of reducing bacteria, the process is the reduction of As(+5) to As(+3). Note that this process is related to the detoxification of the cells. The arsenate ions enter the cells via the phosphate transporters (for example Pst – high affinity phosphate specific transport or Pit – low affinity phosphate inorganic transport), due to structural homologies with phosphate ions. After reaching the cytoplasm, arsenate is reduced into arsenite. This is accomplished by the arsenate-reductase enzyme ArsC or the ArsAB complex. Note that the reduction of arsenate to arsenite is implemented before it is excreted from the cell by the transmembrane protein ArsB. It is mentioned here that ArsB is also known as Acr3 in the context of some eukaryotic micro-organisms). The transformation process followed by the excretion of arsenic is a common occurrence in the living world and is widespread in bacteria [34,39]. Nevertheless, the mobility of arsenic in the natural environment is determined generally by the extent to which it is adsorbed onto the mineral surfaces. Metal oxides tend to be the primary sorbents of As in the environment, especially the oxides of iron (Fe) and aluminum (Al). In addition Mn-oxides can also sorb arsenic to some extent. Among the most important oxides-sinks in nature for the adsorbed, co-precipitated arsenic or even both, are the hydrous ferric, manganese and aluminum oxides. These oxides are composed of octahedrally coordinated metal atoms that share edges to form chains in two and three dimensions structures. However, according to different pH values the OH groups can bind or release H+ ions and this results in the development of a surface charge. In this case arsenic adsorbs this charge by the process of ligand exchange with OH and OH2+ surface functional groups. This yield to the formation of an inner-sphere complex. This type of adsorption requires an incompletely dissociated acid, e.g., H2AsO4-, to provide a proton for the complexation process with the surface OH group so as to form H2O and to providing a space for the anion. The reactions which delineate these geochemical processes are given below [40]: |
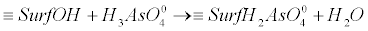 (10) (10) |
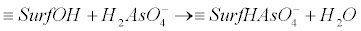 (11) (11) |
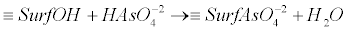 (12) (12) |
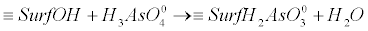 (13) (13) |
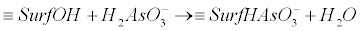 (14) (14) |
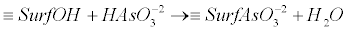 (15) (15) |
| where, ≡SurfOH represents the structural metal atom and associated OH surface functional group. For example ≡SurfHAsO4- is the surface arsenic complex. The energy required to dissociate the weak acid at the oxide surface and the amount of arsenic adsorbed varies with pH. |
| In order to describe adsorption-desorption reactions at the mineral surfaces, two types of models have been proposed. The empirical models are based on the partitioning relationships of a solute between the aqueous and the solid phase, and the conceptual models. The latter models account for the surface complexation and treat adsorption reactions similar to ion association reactions in solution. All models assume that the adsorption reactions are at equilibrium. In the case of the empirical adsorption models the distribution coefficient as in equation (16) is the most known [41]: |
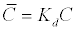 (16) (16) |
| In addition the Langmuir Isotherm and the Freundlich Isotherm are given in equations (17) and (18) respectively [41]. |
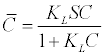 (17) (17) |
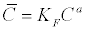 (18) (18) |
The 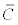 is the adsorbed concentration, C is the aqueous
concentration at equilibrium with the solid phase, S is the adsorption
site concentration, Kd is the distribution coefficient, KL is the Langmuir
equilibrium constant, KF is the Freundlich equilibrium constant, and a
is the Freundlich exponent. is the adsorbed concentration, C is the aqueous
concentration at equilibrium with the solid phase, S is the adsorption
site concentration, Kd is the distribution coefficient, KL is the Langmuir
equilibrium constant, KF is the Freundlich equilibrium constant, and a
is the Freundlich exponent. |
| Arsenic is absorbed by several clay minerals. The maximum adsorption of As(+5) is kaolinite, montmorillonite, illite, halloysite, and chlorite for pH values near 7. The adsorption is decreased with further increase of pH above 7. Adsorption of As(+3) by the same clay minerals is low at low pH values and is increased when pH increases. Arsenate is adsorbed to a greater extent than As(+3) on all clay minerals at pH<7. At higher pH values, adsorption of As(+5) and As(+3) are more comparable, and in some cases As(+3) adsorption exceeds the one of As(+5). Note that only the OH groups that are associated with the Al ions exposed at the edges of clay particles, are considered to be proton acceptors and are able to complex anionic species of As [40,42]. In Table 3 we present selected geo-materials and also minerals, oxides and other constituents that can be use as low-cost effective absorbents for arsenic remediation purposes [43] (Figure 2). The suspension density is 40 g · L-1, and As total concentration is for the single-ion system 20 μM and for the binary system As(+3)T=As(+5)T=20 μM; adopted and modified from [44] (Table 3). |
| Redox fluctuations, pH and ions availability are responsible for the formation of insoluble sulfide precipitates such as arsenopyrite (FeAsS), realgar (AsS), and orpiment (As2S3) in reducing conditions [45]. Arsenic is also found in sedimentary environments, absorbed by Fe(+3) and Mn(+4) oxides – hydroxides after weathering of the sulfide minerals. In the case of the interaction of arsenopyrite with the ferric hydroxide, arsenic is adsorbed or co-precipitated with the ferrosoferric hydroxides via equation (19) [46]: |
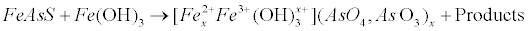 (19) (19) |
| Therefore, high levels of arsenic in natural waters can be due to the reductive dissolution of arsenic rich iron oxyhydroxides [47,48]. In addition oxidative dissolution of arsenic-rich pyrite or arsenopyrite is responsible for As existence in natural waters [49]. In this case, the concentration of Dissolved Oxygen (DO) is the limiting factor for arsenopyrite dissolution in a variety of pH ranges [50,51]. Arsenic release rates seem to increase with increasing DO concentration and temperature, and are similar at low (<7) and high (>10) pH. The reaction includes FeAsS dissolution (equation 20) and As(+3) present as H3AsO3, while Fe(+2) is further oxidized to Fe(OH)3 (equation 21), with an increase of acidity in the solution [52-58]: |
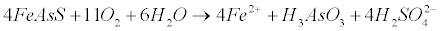 (20) (20) |
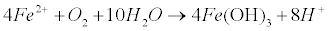 (21) (21) |
| The widespread arsenic contamination is thought to be related with As release from iron oxyhydroxides, probably due to the reaction of Fe-oxides/hydroxides with organic carbon [1,23,59-67]. In such a case the source of As is the adsorbed arsenic onto the surface of Fe-oxides/ hydroxides solid phases, and a parallel release of arsenic during the reductive dissolution of ferric oxides-hydroxides occurs (equation 22) [68-70]: |
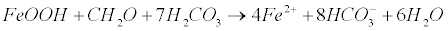 (22) (22) |
| In addition, sorption of As(+3) onto Mn-oxides/hydroxides phases has been reported by Panagopoulos and Panagiotaras [71] in order to delineate controlling geochemical processes in the groundwater pool of the Trifilia karst aquifer, in Western Greece. The proposed mechanism is described by equation (23): |
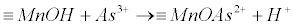 (23) (23) |
| However, adsorbed arsenic species are weak acids and can affect the surface charge due to proton exchange reactions. Whether As adsorbs as a mononuclear or binuclear complex has implications for the level of protonation of the surface species, where this mechanism elucidated in equations (24) and (25) in the case of Fe-oxides/hydroxides surfaces [72-74]: |
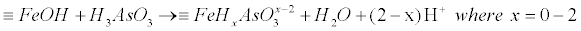 (24) (24) |
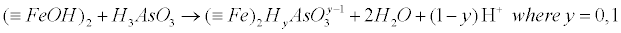 (25) (25) |
| Therefore, further reductive dissolution of ferric oxides-hydroxides by organic matter as described in equation (22), contributes to the cycling of arsenic species into the environment [16]. The overall biogeochemical processes potentially influence arsenic mobility in the natural environment and actually generate specific patterns of distribution and speciation during redox and complexation reactions so that arsenic undergoes a variety of biogeochemical transformations. |
| Conclusion |
| A variety of factors controls the fate of arsenic in the environment. The major biogeochemical transformations of arsenic constituents depended to the primary arsenic source, the redox potential, pH as well as microbial activity. However, the dissolution of arsenic minerals are the major source of the arsenic chemical species in the environment in specific physicochemical conditions. In different environmental conditions arsenic mineral degree of dissolution as well as its chemical species can be established using mathematical modeling. |
| These models can predict the occurrence of different arsenic chemical species throughout redox reactions taking place in specific temperature, pressure and pH conditions. Therefore, arsenopyrite oxidative dissolution is the major geochemical processes regulating the existence of arsenic in natural waters, with the dissolve oxygen concentration being responsible for the degree of dissolution in different pH values. |
| However, Fe-oxides/hydroxides solid phases adsorbed As and they can also release arsenic during their reductive dissolution. In aqueous systems, and in extremely low pH conditions (pH<2), arsenic acid occurs. In addition between pH=2 to pH=11, it is in the form of H2AsO4– and HAsO42- chemical species. As the pH increases from low values, and in mildly reducing conditions, arsenious acid is converting to H2AsO3–. |
| Under oxic conditions Arsenate [As(+5)] is the thermodynamically stable phase of arsenic. In reducing conditions arsenite [As(+3)] ions occurring. The As(+5) chemical species are adsorbed onto hydrous ferric oxides and they also can be released during the microbial reduction of Fe(+3) solid phases. |
| In addition, the mobility and fate of arsenic in the environment is also related to co-precipitation and adsorption onto clay minerals, manganese oxides and hydrous aluminum oxides. In the case of As(+5) ions, maximum adsorption capacities observed in low to up near 7 pH values for chlorite, illite, halloysite, kaolinite and montmorillonite clay minerals. However, adsorption capacity decreases as pH increases. An opposite behavior is apparent for the As(+3) regarding the same clay minerals. In this case adsorption of As(+3) increased with increasing pH, while adsorption was minimum at low pH values. As a concluding remark is that at s, As(+5) ions adsorbed to a greater extent than As(+3) on all the above mentioned clay minerals, while at higher pH values, adsorption of As(+5) and As(+3) were more comparable, and in some cases As(+3) adsorption exceeds that of As(+5). Therefore, clay minerals can be applied for arsenic removal and remediation purposes. |
| Although there is sufficient knowledge on the geochemical processes that governs the arsenic occurrence and fate in nature, the extent to how microbes affecting arsenic fluxes in the environment must be further studied in order to better understand the overall biogeochemical cycling of arsenic in the natural environment. |
| Acknowledgement |
| The authors would like kindly acknowledge the anonymous reviewers for their comments and suggestions helped to improve the quality of the manuscript |
References
|
Tables and Figures at a glance
| Table 1 | Table 2 | Table 3 |
Figures at a glance
 |
 |
| Figure 1 | Figure 2 |
Relevant Topics
- Atmosphere
- Atmospheric Chemistry
- Atmospheric inversions
- Biosphere
- Chemical Oceanography
- Climate Modeling
- Crystallography
- Disaster Science
- Earth Science
- Ecology
- Environmental Degradation
- Gemology
- Geochemistry
- Geochronology
- Geomicrobiology
- Geomorphology
- Geosciences
- Geostatistics
- Glaciology
- Microplastic Pollution
- Mineralogy
- Soil Erosion and Land Degradation
Recommended Journals
Article Tools
Article Usage
- Total views: 20525
- [From(publication date):
April-2015 - Mar 31, 2025] - Breakdown by view type
- HTML page views : 15497
- PDF downloads : 5028