Application of a DNA Deafness Microarray for Detecting Mutations in the Deaf in Vietnam
Received: 09-Apr-2018 / Accepted Date: 16-Apr-2018 / Published Date: 20-Apr-2018 DOI: 10.4172/2155-9872.1000404
Abstract
Object: To identify the presentation of deafness-related gene caused non-syndromic hearing loss in Vietnamese children.
Methods: Apply DNA microarray to 250 hearing-impaired and 250 normal children in Hanoi to screen nine mutational hot spots of four deafness genes, namely GJB2, GJB3, SLC26A4, and 12S rRNA.
Results: there are 16 hearing loss participants carried mutations, account for 6.4% in comparison with 0 % of control group. The carrier rates of GJB2, GJB3, SLC26A4, 12S rRNA mutations were 4.4%, 0%, 1.2% and 0.8% respectively.
Conclusion: Early detection of common deafness mutations is a factor for diagnosing and rescuing, helping hearing-loss children to develop their language and awareness normally. Microarray testing is a helpful and instrumental screening method in the diagnosis of genetic hearing loss.
Keywords: DNA microarray; Non-syndromic hearing loss; GJB2; GJB3; SLC26A4; 12S rRNA
Introduction
Hearing loss (HL) is one of the most common human sensory diseases with an approximate incidence of 1-3 per 1,000 newborns (WHO). Vietnam has about 1 million children born with more than 15,000 children will be deaf each year, according to ASEAN stats 2015. The previous studies showed that 50% or more of congenital hearing loss is due to genetic causes. 70% of all cases of genetic hearing impairment are non-syndromic [1].
To date, more than 200 genes and 300 genetic loci have been implicated in NSHL. The marked heterogeneity of genetic hearing loss can be explained by the complexity of the auditory system, which requires coordination of multiple processes involving the inner ear and nervous system. A defect in any part of this complex chain of events can lead to hearing impairment. The time at which the hearing impairment is discovered is the most important factor affecting the ability of a child to acquire language, because deafness is the biggest obstacle to language learning and communication [2]. Therefore, providers of genetic services should play an increasingly important role in newborn hearing screening, diagnostic, and intervention services.
The most common known cause underlying autosomal recessive NSHL is a mutation in the GJB2 gene. The GJB2 gene encodes connexin 26 (C × 26), a transmembrane protein, functioning in potassium recycling in the inner ear through the formation of gap junctions. GJB2 mutations are responsible for approximately half of the genetic hearing loss cases in the United States, Europe, Israel and China [3-8].
The SLC26A4 gene provides coding instruction for an anion (chloride/ iodide/ bicarbonate) transporter called pendrin, which is expressed in the kidneys, inner ear, and thyroid [9-11]. Mutations in SLC26A4 are responsible for approximately 5-7% of all cases of congenital recessive hearing loss in the East Asian and Caucasian populations [12-14].
The objective of this study is to detect nine hotspot mutations in four common hearing loss related genes, including GJB2 (35delG, 176_191del16, 235delC, 299_300delAT), SLC26A4 (IVS7-2A>G, 2168A>G), GJB3 (538C>T), and 12s rRNA (1555A>G, 1494C>G), through a hereditary hearing loss microarray screening method in Vietnamese children with congenital HL.
Materials and Methods
Research subject
A total of 250 hearing-impaired children at the age from 5 to 8 were recruited from the Otorhinolaryngology faculty and Biomedical and Genetics Department at the Hanoi Medical University, Hanoi, Viet Nam. The control group consisted of 250 subjects without hearing disorder from all corresponding educational grades from 3 primary schools in Hanoi.
Methods
In the study group 500 subjects (cases and control group) were pretested for nine hotspot mutations of 4 deafness genes with a Nine Deafness Gene Mutation Detection Microarray Kit (Capital Bio Corporation, Beijing, China), as previously described.
Firstly, we peripheral blood samples were obtained from objects, and genomic DNA was extracted using a blood genomic DNA extraction kit (Qiagen, America) according to the manufacturer’s protocol. Then we performed polymerase chain reaction (PCR) amplification of the obtained DNA. The PCR product was screened for nine mutations of four deafness-related genes.
Samples were added into 200 μl centrifuge tube and 15 μl of hybridization solution was added. It was placed in the hybridization instrument for 60 min under 50°C and 5 r/min. Then, it was washed, dried and scanned. Deaf Test software was used to set up the QC (quality control probe), PC (positive probe), BC (blank control probe), NC (negative control probe) and W (wild type) and Mutant type) probes for 9 detection sites (Table 1).
| 2D | 7D, 10D | 34D | 21D, 58D | |
|---|---|---|---|---|
| Result |  |
 |
 |
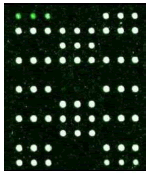 |
| Mutation | GJB2 (299_300delC) Heterozygous |
GJB2 (235delC) Heterozygous |
SLC26A4 (IVS7-2A>G) Heterozygous |
MT-RNR1 (1555A>G) Homozygous |
Table 1: Mutation detection by microarray method.
Results
The carrier and mutation rates of deafness genes by DNA microarray
Among the 250 NSHL cases included in our study, 16 carried at least one type of pathogenic mutation, for a carrier rate of 6.4%. No mutations were identified in the control group (Table 2).
| Gene | Nucleotide chance | Carrier number | Carrier-ate% (n=16) | Carrier rate % in total (n=250) |
|
|---|---|---|---|---|---|
| Homozygous | Heterozygous | ||||
| GJB2 | c.299-300delAT | 3 | 5 | 50% | 3.2% |
| c.235delC | 0 | 3 | 18.75% | 1.2% | |
| 35delG | 0 | 0 | 0% | 0% | |
| 176del16 | 0 | 0 | 0% | 0% | |
| SLC26A4 | c.2168A>G | 0 | 3 | 18.75% | 1.2% |
| IVS7-2A>G | 0 | 0 | 0% | 0% | |
| 12S rRNA | 1555A>G | 2 | 0 | 12.5% | 0.8% |
| 1494C>T | 0 | 0 | 0% | 0% | |
| GJB3 | s538C>T | 0 | 0 | 0% | 0% |
| Total | 16 | 100% | 6.4% | ||
Table 2: The carrier rate of deafness genes in 250 HL children.
Table 1 shows that the c.299-300delAT GJB2 mutation was the most frequent, accounting for 50% of gene mutations. The second most frequent mutation was at c.2168A>G SLC26A4c (18.75%) and c. 235delC GJB2 (18.75%). There are two among the 16 carriers carried the 1555A>G 12S rRNA mutation, account for 2.5%. None of candidates has mutation in GJB3 genes.
Discussion
Several studies show that about 60-80% of pre-lingual HL in patients worldwide is associated with genetic factors, with clinical data from developed countries showing that more than 60% of deaf patients have hereditary deafness [15]. As the human genome project had been completed, deaf disease gene localization and cloning have made great progress. The molecular genetics and molecular epidemiology of NSHL have gradually revealed the underlying genetic susceptibility to HL.
Our new findings revealed that in the 250 patients screened, a total of 16 of these subjects had pathogenic mutations, accounting for 6.4%, which is in accordance with the frequencies in previous reports. Among these nine sites in four genes, the carrier rate of GJB2 mutations (235 del C, 299 del AT. 35 del G, 176 del 16) were 3.2%, 1.2%, 0%, and 0%, respectively. The mutation rate for SLC26A4 sites (IVS7-2A>G, 2168 A>G) was 1.2% and 0%, respectively. The figure of gene MT-RNR1 (m.1555A>G) was 0.8% and none of mutation in GJB3 was found.
GJB2 gene mutations are the most common deafness-susceptibility genes, which already have been mapped and cloned. At present, more than 100 types of mutations have been found in the GJB2 gene. However, different populations differ in their variations in the GJB2 gene [16]. Typically, the audiogram has a down sloping or flat pattern. Symmetry between ears is typical, although one-fourth of individuals have intra-aural differences of up to 20 dB (Cohn and Kelley). The loss tends to be stable, with neither improvement in hearing nor fluctuation in hearing level over the long term. In general, bony abnormalities of the cochlea are not part of the deafness phenotype (less than 10%) and developmental motor milestones and vestibular function are normal.
SLC26A4 encodes an anion transporter (chloride and iodide) and mutations in SLC26A4 are the second most frequent cause of autosomal recessive nonsyndromic HL (ARNSHL), and the resulting phenotypes include PS, an autosomal recessive disorder characterized by sensorineural deafness and goiter, Everett et al. [9]. The deafness is congenital and associated with temporal bone abnormalities that range in severity from isolated enlargement of the vestibular aqueduct [EVA; dilated vestibular aqueduct (DVA)] to Mondini dysplasia, a more complex malformation that also includes cochlear hypoplasia. The thyromegaly in PS is due to multinodular goitrous changes in the thyroid gland, although affected persons typically remain euthyroid. The perchlorate discharge test is often abnormal.
MT-RNR1 is a drug-sensitive mutation; knowing about this mutation can help parents avoid drugs, which will adversely affect the child’s hearing. Deafness genes identified at neonatal screening can predict some genetic correlation of late-onset deafness.
GJB3 mutation is associated with the high-frequency hearing impairment in an autosomal dominant or recessive inheritance pattern. GJB3 mutation is rarely detected in European and North American population [17]. Our result has shown that there is no mutation of gen GJB3 in Vietnam.
Conclusion
In conclusion, about 6.4% of the children with severe or profound NSHL were found to harbor nine common mutations in the GJB2, SLC26A4, MT-RNR1. This study supports the theory that the mutations of GJB2 is the major genetic causes in sporadic NSHL, which accounts for approximately 68.75% of case.
Hopefully a more comprehensive, accurate, fast, economic gene chip will be developed in near future. As a result, deafness detection technique will be used as a routine detection in clinic.
References
- Smith RJ, Bale JF, White KR (2005) Sensorineural hearing loss in children. Lancet 368: 879-890.
- Pimperton H, Kennedy CR (2012) The impact of early identification of permanent childhood hearing impairment on speech and language outcomes. Arch Dis Child 97: 648-653.
- Estivill X, Fortina P, Surrey S, Rabionet R, Melchionda S, et al. (1998) Connexin-26 mutations in sporadic and inherited sensorineural deafness. Lancet 351: 394-398.
- Morell RJ, Kim HJ, Hood LJ, Goforth L, Friderici K, et al. (1998) Mutations in the connexin 26 gene (GJB2) among Ashkenazi Jews with non-syndromic recessive deafness. N Engl J Med 339: 1500-1505.
- Kelley PM, Harris DJ, Comer BC, Askew JW, Fowler T, et al. (1998) Novel mutations in the connexin 26 gene (GJB2) that cause autosomal recessive (DFNB1) hearing loss. Am J Hum Genet 62: 792-799.
- Scott DA, Kraft ML, Stone EM, Sheffield VC, Smith RJ (1998) Connexin mutations and hearing loss. Nature 391: 32.
- Brobby GW, Muller-Myhsok B, Horstmann RD (1998) Connexin 26 R143W mutation associated with recessive nonsyndromic sensorineural deafness in Africa. N Engl J Med 338: 548-550.
- Yuan Y, You Y, Huang D, Cui J, Wang Y, et al. (2009) Comprehensive molecular etiology analysis of nonsyndromic hearing impairment from typical areas in China. J Transl Med 7: 79.
- Everett LA, Morsli H, Wu DK, Green ED (1999) Expression pattern of the mouse ortholog of the Pendred’s syndrome gene (Pds) suggests a key role for pendrin in the inner ear. Proc Natl Acad Sci USA 96: 9727-9732.
- Royaux IE, Suzuki K, Mori A, Katoh R, Everett LA, et al. (2000) Pendrin, the protein encoded by the Pendred syndrome gene (PDS), is an apical porter of iodide in the thyroid and is regulated by thyroglobulin in FRTL-5 cells. Endocrinology 141: 839-845.
- Mount DB, Romero MF (2004) The SLC26 gene family of multifunctional anion exchangers. Pflugers Arch 447: 710-721.
- Park HJ, Shaukat S, Liu XZ, Hahn SH, Naz S, et al. (2003) Origins and frequencies of SLC26A4 (PDS) mutations in east and south Asians: global implications for the epidemiology of deafness. J Med Genet 40: 242-248.
- Hutchin T, Coy NN, Conlon H, Telford E, Bromelow K, et al. (2005) Assessment of the genetic causes of recessive childhood nonsyndromic deafness in the UK–implications for genetic testing. Clin Genet 68: 506-512.
- Anwar S, Riazuddin S, Ahmed ZM, Tasneem S, Ateeq-ul-Jaleel, et al. (2009) SLC26A4 mutation spectrum associated with DFNB4 deafness and Pendred’s syndrome in Pakistanis. J Hum Genet 54: 266-270.
- Hilgert N, Smith RJH, Van Camp G (2009) Forty-six genes causing nonsyndromic hearing impairment: which ones should be analyzed in DNA diagnostics. Mutat Res Rev Mutat Res 681: 189-196.
- Liu X, Xia X, Ke X, Ouyang XM, Du LL, et al. (2002) The prevalence of connexin 26 (GJB2) mutations in the Chinese population. Hum Genet 111: 394-397.
- Frei K, Ramsebner R, Hamader G, Lucas T, Schoefer C, et al. (2004) Lack of association between Connexin 31 (GJB3) alterations and sensorineural deafness in Austria. Hear Res 194: 81-86.
Citation: Trang NT, Giang VT (2018) Application of a DNA Deafness Microarray for Detecting Mutations in the Deaf in Vietnam. J Anal Bioanal Tech 9: 404. DOI: 10.4172/2155-9872.1000404
Copyright: © 2018 Trang NT, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Share This Article
Open Access Journals
Article Tools
Article Usage
- Total views: 4758
- [From(publication date): 0-2018 - Apr 04, 2025]
- Breakdown by view type
- HTML page views: 3889
- PDF downloads: 869