|
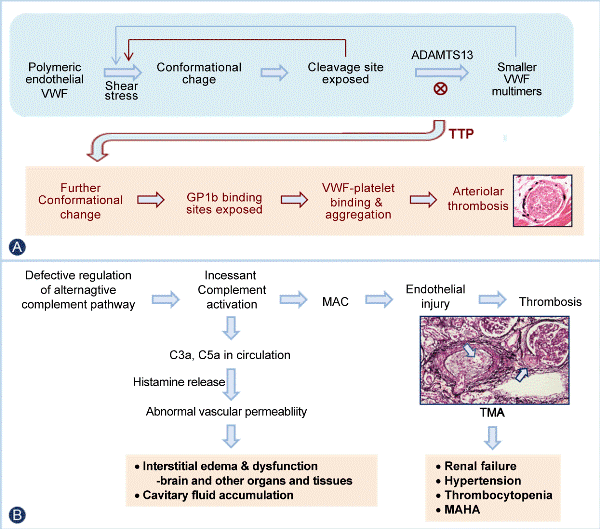
| Figure 1: Pathogenesis of thrombosis in TTP and of TMA in aHUS. A. In normal circulation, VWF, secreted from endothelial cells as a polymeric form, is cleaved by ADAMTS13 to become progressively small multimers whenever its conformation is beginning to be unfolded by high levels of shear stress in arterioles and capillaries. This design ensures that large forms of VWF are present in the circulation and readily available for hemostasis at sites of vessel injury, while it keeps VWF multimers in compact, inactive conformation to prevent platelet thrombosis in the circulation. When ADAMTS13 is deficient, VWF, after repeated cycles of conformational change by shear stress, becomes unfolded and activated to cause platelet aggregation, resulting in VWF-platelet thrombosis in capillaries and arterioles characteristic of TTP. B. In aHUS, in which regulation of the alternative complement pathway is defective, activation of the complement system, either spontaneous or triggered, results in incessant generation of complement activation products, including membrane attack complex (MAC, i.e. C5b9), which causes thrombotic microangiopathy (TMA) with endothelial injury (light-blue arrow pointing to arteriolar stenosis due to expansion of subendothelium) and thrombosis (dark blue arrow pointing to thrombosis in a juxtaglomerular arteriole) in the kidney; and anaphylatoxins C3a and C5a, which are released in the circulation and induces the secretion of histamine and perhaps other vasoactive mediators from basophils and mast cells in the brain and other organs. Together these processes cause, in addition to thrombocytopenia and microangiopathic hemolysis, renal failure, hypertension, interstitial edema, cavitary fluid accumulation, and dysfunction of multiple organs characteristic of aHUS. |