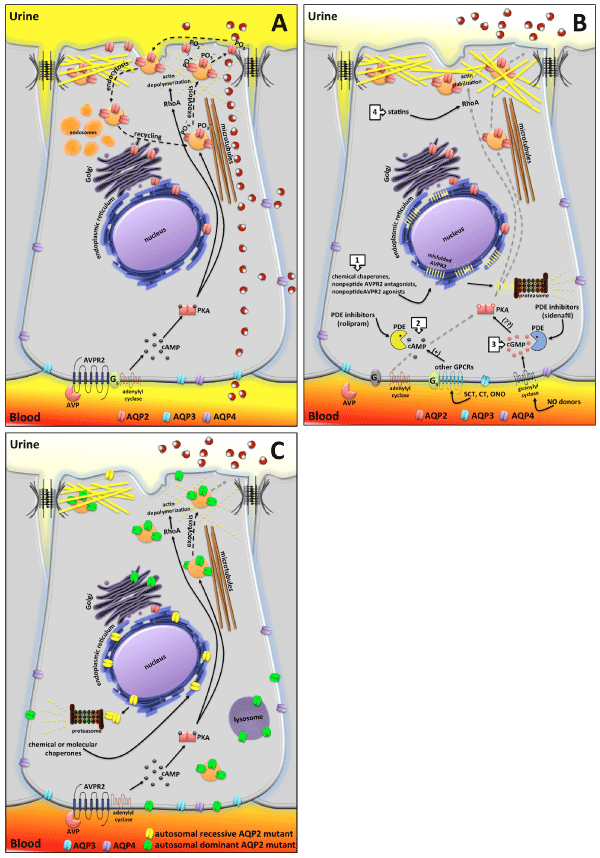
B) AVPR2 mutations are responsible of X-linked NDI. The most recurrent mutations of AVPR2 responsible of X-linked NDI are class II mutations encompassing missense or insertion/deletion producing full-length misfolded proteins often retaining intrinsic functionality but trapped in the endoplasmic reticulum and mostly targeted for proteasome degradation. Lack of plasma membrane expressed AVPR2 prevents AVP signaling and AQP2 exocytosis. Unconventional strategies for treating X-linked NDI are focused on: 1) use of chemical chaperones aiding protein folding and inducing export from the ER; nonpeptide AVPR2 antagonists, like vaptans, that stabilize receptor conformation in the ER, thus allowing escape from the ER quality control mechanism; nonpeptide AVPR2 agonists that interact intracellularly with pre-formed receptor-G proteinadenylate cyclase (AC) complexes, thus increasing cAMP concentration. 2) activation of the cAMP pathway by stimulating other G proteins-coupled receptors (GPCRs) coupled to Gs/adenylyl cyclase expressed in collecting duct principal cells; inhibition of phosphodiestherases (PDE) to increase basal cAMP levels. 3) activation of the cGMP pathway promoting AQP2 exocytosis either by stimulating guanylyl cyclase or by inhibiting PDE to increase basal cGMP levels. 4) statins treatment that, by inhibiting prenylation of RhoA, facilitates cortical actin depolymerization and constitutive exocytosis of AQP2 at the apical membrane.
C) AQP2 mutations explain autosomal recessive and dominant NDI. Most of the AQP2 mutations falling in the protein transmembrane domains are misfolded (yellow tetramers) and retained in the ER until degraded in the proteasome. Affected patients are homozygous or compound heterozygous for these AQP2 mutations. Since most of these mutants still maintain water channels functionality, a therapeutic approach under investigation is based on the use of chemical chaperones aiding release from the ER. Autosomal dominant NDI is caused by AQP2 mutations affecting the COOH-terminus of the protein, which is a crucial domain for phosphorylation or apical sorting. These AQP2 mutants have a dominant effect over wtAQP2 subunit and are responsible of AQP2 missorting. See text for more detailed explanation.