|
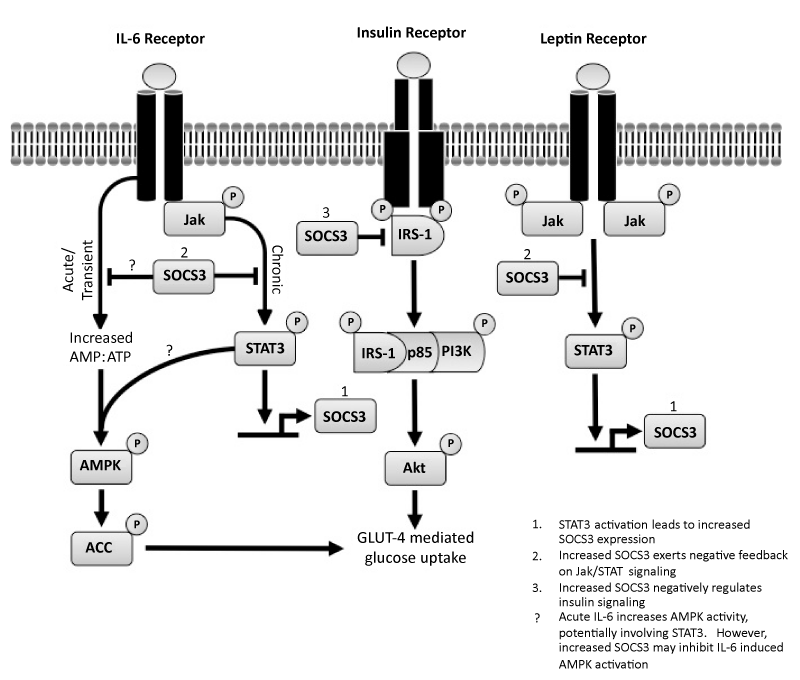
| Figure 1: IL-6, insulin, and leptin signaling in skeletal muscle. Chronic low-grade inflammation is associated with increased levels of IL-6, which leads to increased phosphorylation of STAT3, and subsequent increased SOCS3 expression. Increased SOCS3 mediates the inhibitory effects of IL-6 on insulin signaling and glucose uptake. SOCS3 directly disrupts insulin signaling by binding to IR and preventing interaction with IRS-1 (primarily in skeletal muscle and adipose tissue) / IRS-2 (primarily in the liver), and may target IRS-1/IRS-2 for degradation. Disrupting the early signaling events also has downstream effects on other proteins in the insulin pathway including PI3K and Akt, ultimately leading to insulin resistance and attenuated GLUT-4 mediated glucose uptake. Chronically elevated leptin levels also leads to increased SOCS3 expression, which can also directly inhibit insulin mediated glucose uptake. In addition, SOCS3 can have indirect effects on glucose uptake; increased SOCS3 leading to leptin resistance, and impaired leptin mediated AMPK activation. In contrast, acute/transient IL-6 elevations in response to exercise do not cause sustained increased SOCS3 expression, but cause increased insulin sensitivity in peripheral tissues by increased AMPK activity and subsequent increased glucose uptake. IL-6 mediated activation of AMPK in response to exercise is blunted in chronic inflammatory states. Since IL-6 is required for exercise-induced AMPK activation and obesity results in a blunted exercise-induced activation of AMPK, it is possible that increased SOCS3 may cause IL-6 resistance, which results in diminished AMPK activity and increased insulin resistance. IL-6: Interleukin-6; STAT3: Signal Transducer and Activator of Transcription 3; SOCS3: Suppressor of Cytokine Signaling 3; IR: Insulin Receptor; IRS: Insulin Receptor Substrate; PI3K: Phosphoinositide 3 Kinase; GLUT-4: Glucose Transporter 4; AMPK: AMP activated Protein Kinase |